
In Vivo Reversal of P-Glycoprotein-Mediated Drug Resistance in a Breast Cancer Xenograft and in Leukemia Models Using a Novel, Potent, and Nontoxic Epicatechin EC31

 ,
,
Abstract
:1. Introduction
2. Results
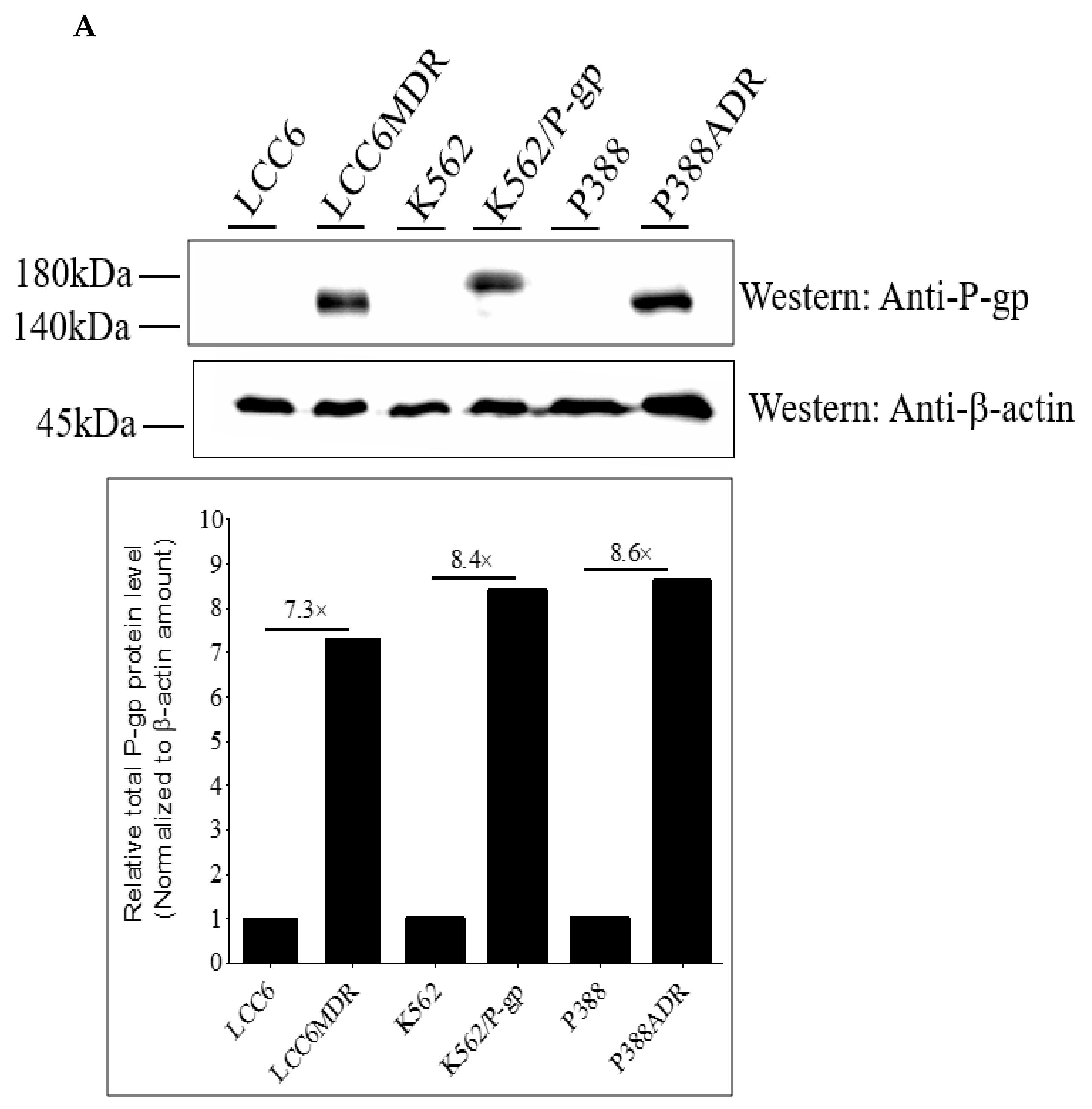
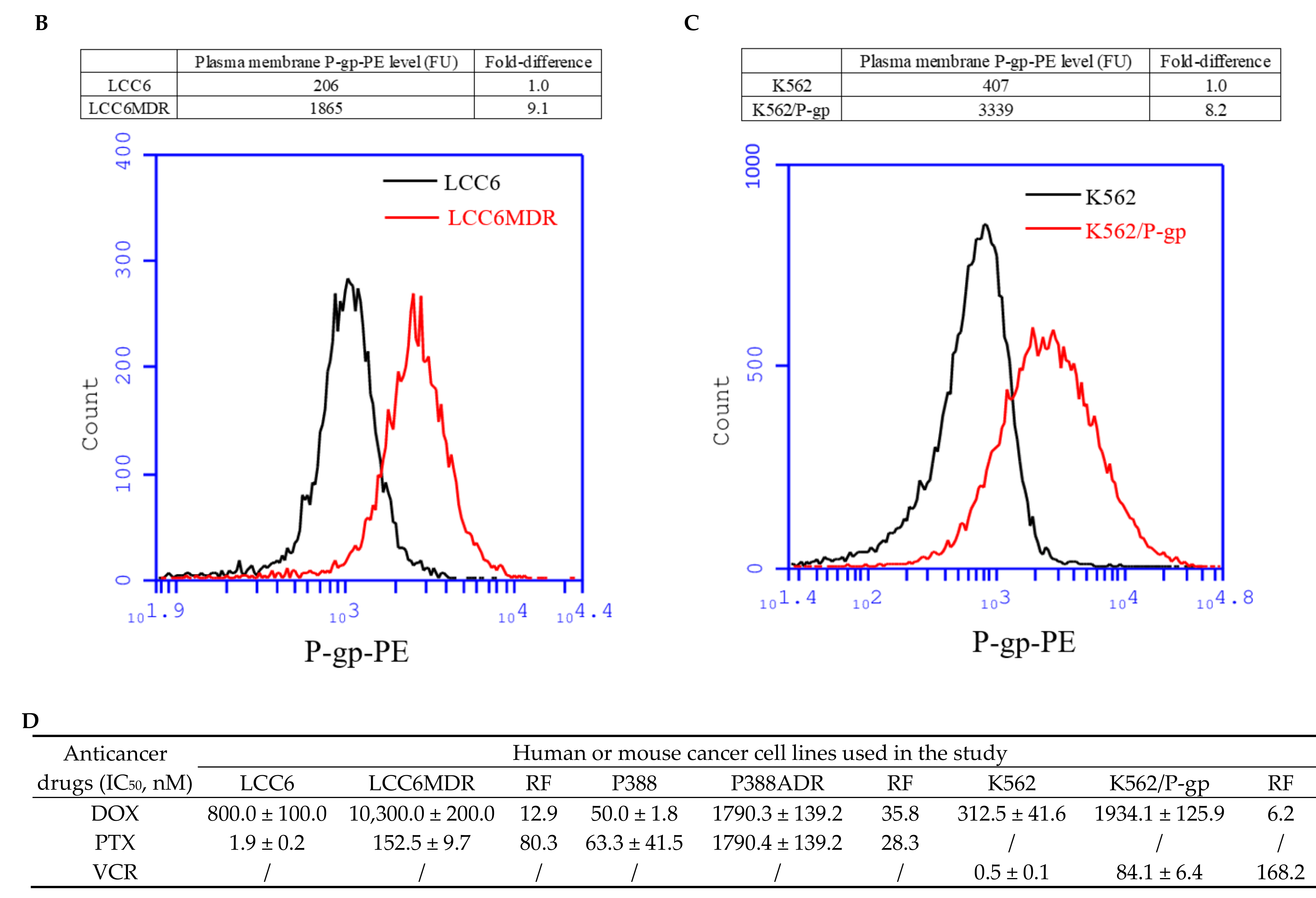
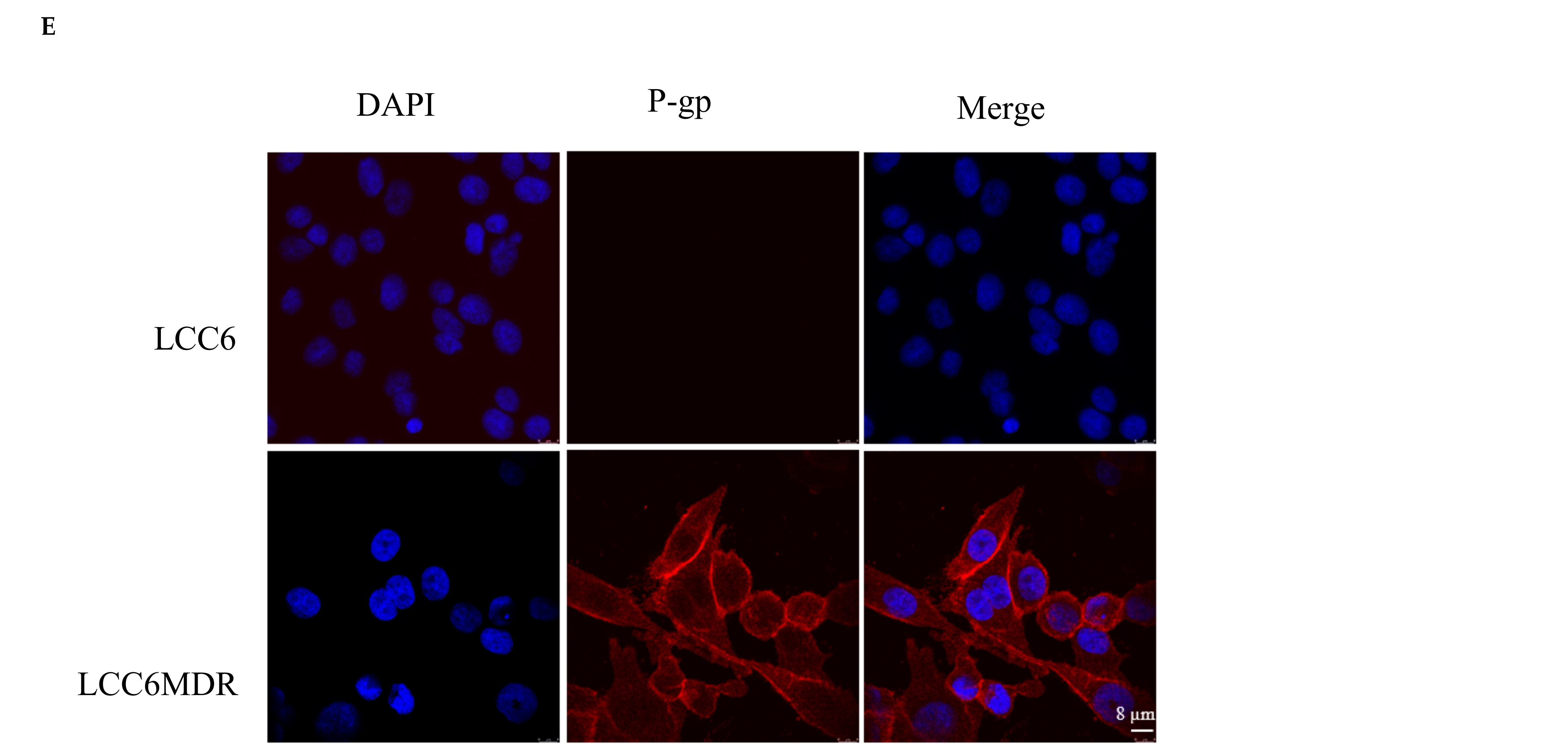
2.1. Methylated Catechin Derivatives Modulated P-gp-Mediated MDR In Vitro
2.2. Methylated Catechin Derivatives Increased DOX Accumulation by Inhibiting the Transport Activity of P-gp
2.3. Methylated EC31 and C25 Did Not Inhibit P-gp ATPase Activity
2.4. Methylated EC31 Was Not a P-gp Transport Substrate
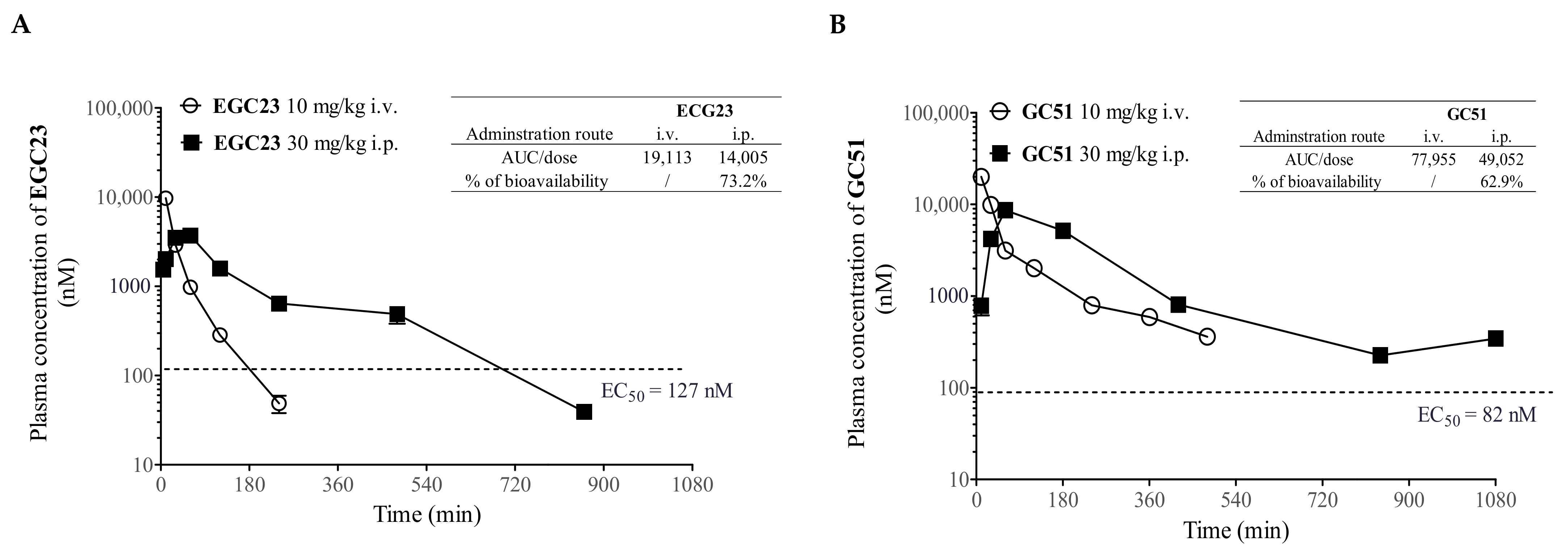
2.5. Pharmacokinetic (PK) Study of Methylated Catechin Derivatives and Their Effects on the PK of PTX in Mice
2.6. EC31 Alone and Its Combination with PTX Did Not Induce Toxicity in BALB/c Mice
2.7. EC31 Increased the Intratumor PTX Concentration and Reversed the P-gp-Mediated PTX Resistance in the LCC6MDR Tumor Xenograft Model
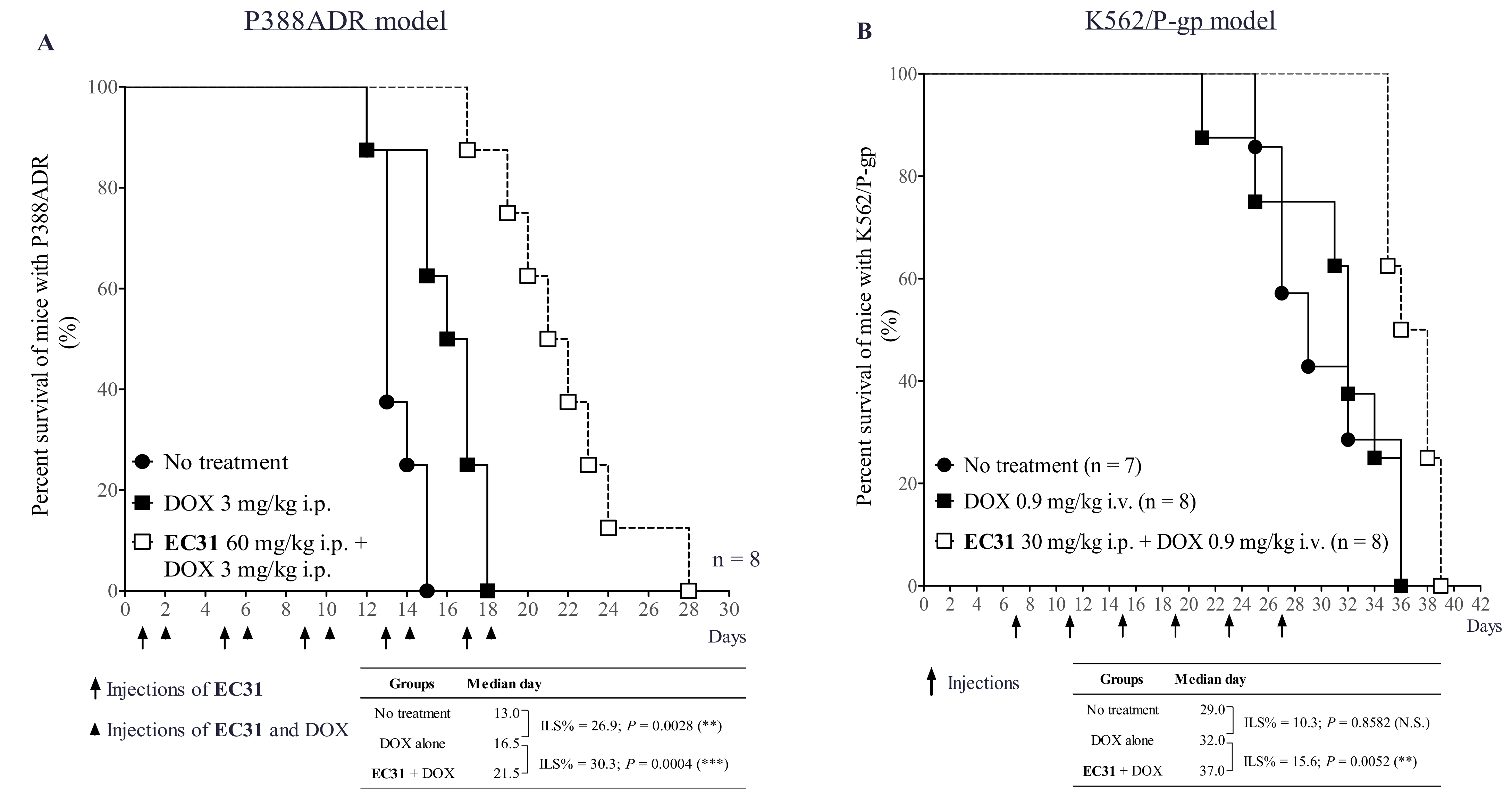
2.8. EC31 Reversed the DOX Resistance in the Murine Leukemia P388ADR and Human Leukemia K562/P-gp Models
3. Discussion
4. Materials and Methods
4.1. Catechin Analogues and Cell Lines
4.2. Cell Cultures
4.3. In Vitro Cell Proliferation Assay
4.4. DOX Accumulation and Efflux Assays
4.5. Determination of Total P-gp Expression Using Western Blot
4.6. Determination of Plasma Membrane P-gp Level Using Flow Cytometer
4.7. Determination of P-gp Cellular Localization Using Immunofluorescence Staining
4.8. P-gp ATPase Activity Assay
4.9. UPLC-MS/MS Detection of Methylated Catechin Derivatives and PTX
4.10. Intracellular EC31 Accumulation and Efflux
4.11. Pharmacokinetic Study of Methylated Catechin Derivatives and PTX in Plasma
4.12. In Vivo Toxicity Study of EC31 and Its Combination with PTX
4.13. In Vivo Efficacy of EC31 in Reversing PTX Resistance in LCC6MDR Tumor Xenograft Model
4.14. In Vivo PTX and EC31 Accumulation in Tumor
4.15. In Vivo Efficacy of EC31 in Reversing DOX Resistance in Murine Leukemia P388ADR Model
4.16. In Vivo Efficacy of EC31 in Reversing DOX Resistance in Human Leukemia K562/P-gp Model
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, A.; Ierano, C.; Szakacs, G.; Robey, R.W.; Bates, S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar] [PubMed]
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef]
- Hait, W.N.; Yang, J.M. Clinical management of recurrent breast cancer: Development of multidrug resistance (MDR) and strategies to circumvent it. Semin. Oncol. 2005, 32 (Suppl. 7), S16–S21. [Google Scholar] [CrossRef]
- Punyammalee, B.; Manoromana, S.; Purisa, W.; Chariyalertsak, S.; Rerkamnuaychok, B. Association of mdr1 gene expression with other prognostic factors and clinical outcome in human breast cancer. J. Med. Assoc. Thai. 1997, 80 (Suppl. 1), S162–S173. [Google Scholar]
- Komdeur, R.; Plaat, B.E.; van der Graaf, W.T.; Hoekstra, H.J.; Hollema, H.; van den Berg, E.; Zwart, N.; Scheper, R.J.; Molenaar, W.M. Expression of multidrug resistance proteins, P-gp, MRP1 and LRP, in soft tissue sarcomas analysed according to their histological type and grade. Eur. J. Cancer 2003, 39, 909–916. [Google Scholar] [CrossRef]
- Plaat, B.E.; Hollema, H.; Molenaar, W.M.; Torn Broers, G.H.; Pijpe, J.; Mastik, M.F.; Hoekstra, H.J.; van den Berg, E.; Scheper, R.J.; van der Graaf, W.T. Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: Differences in clinical outcome and expression of multidrug resistance proteins. J. Clin. Oncol. 2000, 18, 3211–3220. [Google Scholar] [CrossRef]
- Li, J.; Xia, X.; Hua, D.; Wang, W.; Chen, Z.; Ruan, C. Clinical and biological relevance of flow cytometric determination of P-glycoprotein expression in acute non-lymphocytic leukemia. Chin. Med. J. 1997, 110, 919–922. [Google Scholar]
- Rao, D.N.; Anuradha, C.; Vishnupriya, S.; Sailaja, K.; Surekha, D.; Raghunadharao, D.; Rajappa, S. Association of an MDR1 gene (C3435T) polymorphism with acute leukemia in India. Asian Pac. J. Cancer Prev. 2010, 11, 1063–1066. [Google Scholar]
- Willman, C.L. The prognostic significance of the expression and function of multidrug resistance transporter proteins in acute myeloid leukemia: Studies of the Southwest Oncology Group Leukemia Research Program. Semin. Hematol. 1997, 34 (Suppl. 5), 25–33. [Google Scholar] [PubMed]
- Czyzewski, K.; Styczynski, J. Imatinib is a substrate for various multidrug resistance proteins. Neoplasma 2009, 56, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litman, T.; Druley, T.E.; Stein, W.D.; Bates, S.E. From MDR to MXR: New understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol. Life Sci. 2001, 58, 931–959. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Friedenberg, W.R.; Rue, M.; Blood, E.A.; Dalton, W.S.; Shustik, C.; Larson, R.A.; Sonneveld, P.; Greipp, P.R. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): A trial of the Eastern Cooperative Oncology Group. Cancer 2006, 106, 830–838. [Google Scholar] [CrossRef]
- Baer, M.R.; George, S.L.; Dodge, R.K.; O'Loughlin, K.L.; Minderman, H.; Caligiuri, M.A.; Anastasi, J.; Powell, B.L.; Kolitz, J.E.; Schiffer, C.A.; et al. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood 2002, 100, 1224–1232. [Google Scholar] [CrossRef]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther. 2007, 7, 447–459. [Google Scholar] [CrossRef]
- Cripe, L.D.; Uno, H.; Paietta, E.M.; Litzow, M.R.; Ketterling, R.P.; Bennett, J.M.; Rowe, J.M.; Lazarus, H.M.; Luger, S.; Tallman, M.S. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: A randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood 2010, 116, 4077–4085. [Google Scholar] [CrossRef]
- Tang, R.; Faussat, A.M.; Perrot, J.Y.; Marjanovic, Z.; Cohen, S.; Storme, T.; Morjani, H.; Legrand, O.; Marie, J.P. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer 2008, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Shien, K.; Suzawa, K.; Tsukuda, K.; Tomida, S.; Sato, H.; Torigoe, H.; Watanabe, M.; Namba, K.; Yamamoto, H.; et al. Elacridar, a third-generation ABCB1 inhibitor, overcomes resistance to docetaxel in non-small cell lung cancer. Oncol. Lett. 2017, 14, 4349–4354. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, K.; Thangapandian, S.; Tajkhorshid, E. Extended-ensemble docking to probe dynamic variation of ligand binding sites during large-scale structural changes of proteins. Chem. Sci. 2022, 13, 4150–4169. [Google Scholar] [CrossRef]
- Safa, A.R. Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr. Med. Chem. Anticancer Agents 2004, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, P.L.; Lee, S.J.; Advani, R.; Tallman, M.S.; Sikic, B.I.; Letendre, L.; Dugan, K.; Lum, B.; Chin, D.L.; Dewald, G.; et al. Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: A phase III trial (E2995). J. Clin. Oncol. 2004, 22, 1078–1086. [Google Scholar] [CrossRef]
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.F.; Pokharel, D.; Bebawy, M. MRP1 and its role in anticancer drug resistance. Drug Metab. Rev. 2015, 47, 406–419. [Google Scholar] [CrossRef]
- Jodoin, J.; Demeule, M.; Beliveau, R. Inhibition of the multidrug resistance P-glycoprotein activity by green tea polyphenols. Biochim. Biophys. Acta 2002, 1542, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Wong, I.L.; Wang, B.C.; Yuan, J.; Duan, L.X.; Liu, Z.; Liu, T.; Li, X.M.; Hu, X.; Zhang, X.Y.; Jiang, T.; et al. Potent and Nontoxic Chemosensitizer of P-Glycoprotein-Mediated Multidrug Resistance in Cancer: Synthesis and Evaluation of Methylated Epigallocatechin, Gallocatechin, and Dihydromyricetin Derivatives. J. Med. Chem. 2015, 58, 4529–4549. [Google Scholar] [CrossRef]
- Wong, I.L.K.; Wang, X.K.; Liu, Z.; Sun, W.; Li, F.X.; Wang, B.C.; Li, P.; Wan, S.B.; Chow, L.M.C. Synthesis and evaluation of stereoisomers of methylated catechin and epigallocatechin derivatives on modulating P-glycoprotein-mediated multidrug resistance in cancers. Eur. J. Med. Chem. 2021, 226, 113795. [Google Scholar] [CrossRef]
- Han, L.W.; Gao, C.; Mao, Q. An update on expression and function of P-gp/ABCB1 and BCRP/ABCG2 in the placenta and fetus. Expert Opin. Drug Metab. Toxicol. 2018, 14, 817–829. [Google Scholar] [CrossRef]
- Yan, C.S.; Wong, I.L.; Chan, K.F.; Kan, J.W.; Chong, T.C.; Law, M.C.; Zhao, Y.; Chan, S.W.; Chan, T.H.; Chow, L.M. A New Class of Safe, Potent, and Specific P-gp Modulator: Flavonoid Dimer FD18 Reverses P-gp-Mediated Multidrug Resistance in Human Breast Xenograft In Vivo. Mol. Pharm. 2015, 12, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, N.; Tsutsui, Y.; Tagashira, T.; Choshi, T.; Hibino, S.; Kamishikiryou, J.; Furuno, K. The ability of gallate and pyrogallol moieties of catechins to inhibit P-glycoprotein function. J. Functional. Food 2011, 3, 298–304. [Google Scholar] [CrossRef]
- Yusa, K.; Tsuruo, T. Reversal mechanism of multidrug resistance by verapamil: Direct binding of verapamil to P-glycoprotein on specific sites and transport of verapamil outward across the plasma membrane of K562/ADM cells. Cancer Res. 1989, 49, 5002–5006. [Google Scholar] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Detty, M.R.; Clarke, D.M. The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J. Biol. Chem. 2012, 287, 26806–26816. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Clarke, D.M. Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem. Pharmacol. 2014, 92, 558–566. [Google Scholar] [CrossRef]
- Ritchie, T.K.; Kwon, H.; Atkins, W.M. Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J. Biol. Chem. 2011, 286, 39489–39496. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.F.; Wong, I.L.; Kan, J.W.; Yan, C.S.; Chow, L.M.; Chan, T.H. Amine linked flavonoid dimers as modulators for P-glycoprotein-based multidrug resistance: Structure-activity relationship and mechanism of modulation. J. Med. Chem. 2012, 55, 1999–2014. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Chemical Structures | Cpds at 1 μM | |||||
|---|---|---|---|---|---|---|
| DMSO | EGCG (10 μM) | EGC23 a | GC51 a | EC31 b | C25 b | |
| R1 | / | H | Me | Me | Me | Me |
| R2 | / | OH | OMe | OMe | H | H |
| Linker | / | / |  |  |  |  |
| Position C2 | / | R | R | R | R | R |
| Position C3 | / | R | R | S | R | S |
| LCC6MDR P-gp-modulating activity Mean IC50 of PTX (nM) [RF] | 152.5 b [1.0] | 122.6 a [1.2] | 3.7 a [41.2] | 2.9 a [52.6] | 2.2 b [69.3] | 1.8 b [84.7] |
| HEK293/R2 BCRP-modulating activity Mean IC50 of topotecan (nM) [RF] | 295.6 b [1.0] | ND | 357.0 a [ 0.8] | 38.0 a [7.8] | 100.8 b [2.9] | 45.5 b [6.5] |
| 2008MRP1 MRP1-modulating activity Mean IC50 of DOX (nM) [RF] | 426.5 b [1.0] | ND | 322.0 a [1.3] | 141.0 a [ 3.0] | 341.0 b [1.3] | 353.7 b [1.2] |
| Cpds | EC50 (nM) for Reversing MDR | |||||
|---|---|---|---|---|---|---|
| DOX Resistance | PTX Resistance | VCR Resistance | ||||
| LCC6MDR | P388ADR | K562/P-gp | LCC6MDR | P388ADR | K562 | |
| EGC23 | 244 ± 23 | 385 ± 16 | 165 ± 22 | 127 ± 30 | 168 ± 47 | 107 ± 6 |
| GC51 | 233 ± 36 | 335 ± 15 | 129 ± 36 | 82 ± 14 | 496 ± 12 | 77 ± 7 |
| EC31 | 159 ± 21 | 186 ± 31 | 93 ± 33 | 94 ± 21 | 249 ± 18 | 37 ± 10 |
| C25 | 180 ± 18 | 260 ± 60 | 93 ± 21 | 91 ± 5 | 216 ± 10 | 60 ± 12 |
| EGCG | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, W.; Wong, I.L.K.; Law, H.K.-W.; Su, X.; Chan, T.C.F.; Sun, G.; Yang, X.; Wang, X.; Chan, T.H.; Wan, S.; et al. In Vivo Reversal of P-Glycoprotein-Mediated Drug Resistance in a Breast Cancer Xenograft and in Leukemia Models Using a Novel, Potent, and Nontoxic Epicatechin EC31. Int. J. Mol. Sci. 2023, 24, 4377. https://doi.org/10.3390/ijms24054377
Sun W, Wong ILK, Law HK-W, Su X, Chan TCF, Sun G, Yang X, Wang X, Chan TH, Wan S, et al. In Vivo Reversal of P-Glycoprotein-Mediated Drug Resistance in a Breast Cancer Xenograft and in Leukemia Models Using a Novel, Potent, and Nontoxic Epicatechin EC31. International Journal of Molecular Sciences. 2023; 24(5):4377. https://doi.org/10.3390/ijms24054377
Chicago/Turabian StyleSun, Wenqin, Iris L. K. Wong, Helen Ka-Wai Law, Xiaochun Su, Terry C. F. Chan, Gege Sun, Xinqing Yang, Xingkai Wang, Tak Hang Chan, Shengbiao Wan, and et al. 2023. "In Vivo Reversal of P-Glycoprotein-Mediated Drug Resistance in a Breast Cancer Xenograft and in Leukemia Models Using a Novel, Potent, and Nontoxic Epicatechin EC31" International Journal of Molecular Sciences 24, no. 5: 4377. https://doi.org/10.3390/ijms24054377
APA StyleSun, W., Wong, I. L. K., Law, H. K. -W., Su, X., Chan, T. C. F., Sun, G., Yang, X., Wang, X., Chan, T. H., Wan, S., & Chow, L. M. C. (2023). In Vivo Reversal of P-Glycoprotein-Mediated Drug Resistance in a Breast Cancer Xenograft and in Leukemia Models Using a Novel, Potent, and Nontoxic Epicatechin EC31. International Journal of Molecular Sciences, 24(5), 4377. https://doi.org/10.3390/ijms24054377