
Mesenchymal Stem Cells in Acquired Aplastic Anemia: The Spectrum from Basic to Clinical Utility




Abstract
:1. Introduction
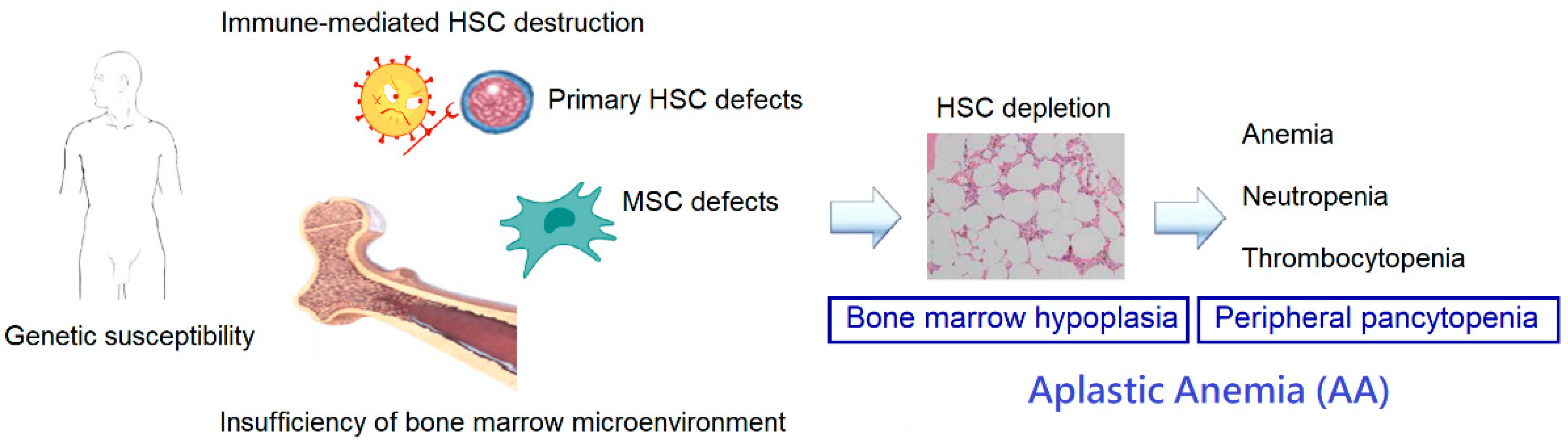
2. Pathophysiology of Aplastic Anemia
2.1. Immune Dysfunction
2.2. Deficiencies of HSCs
2.3. Genetic Susceptibility
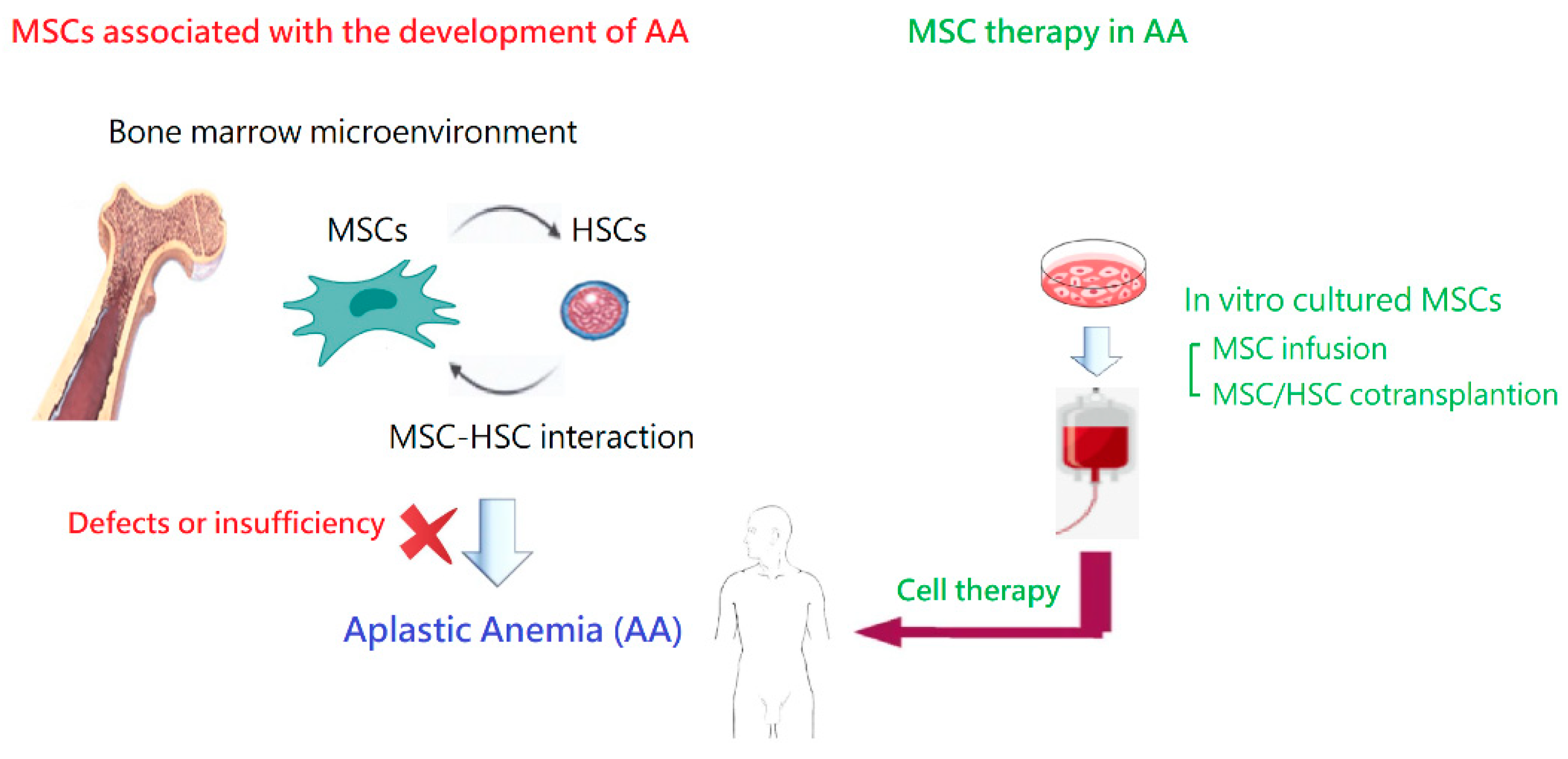
2.4. Alterations in the Bone Marrow Microenvironment
3. Mesenchymal Stem Cells
3.1. MSCs in the Bone Marrow
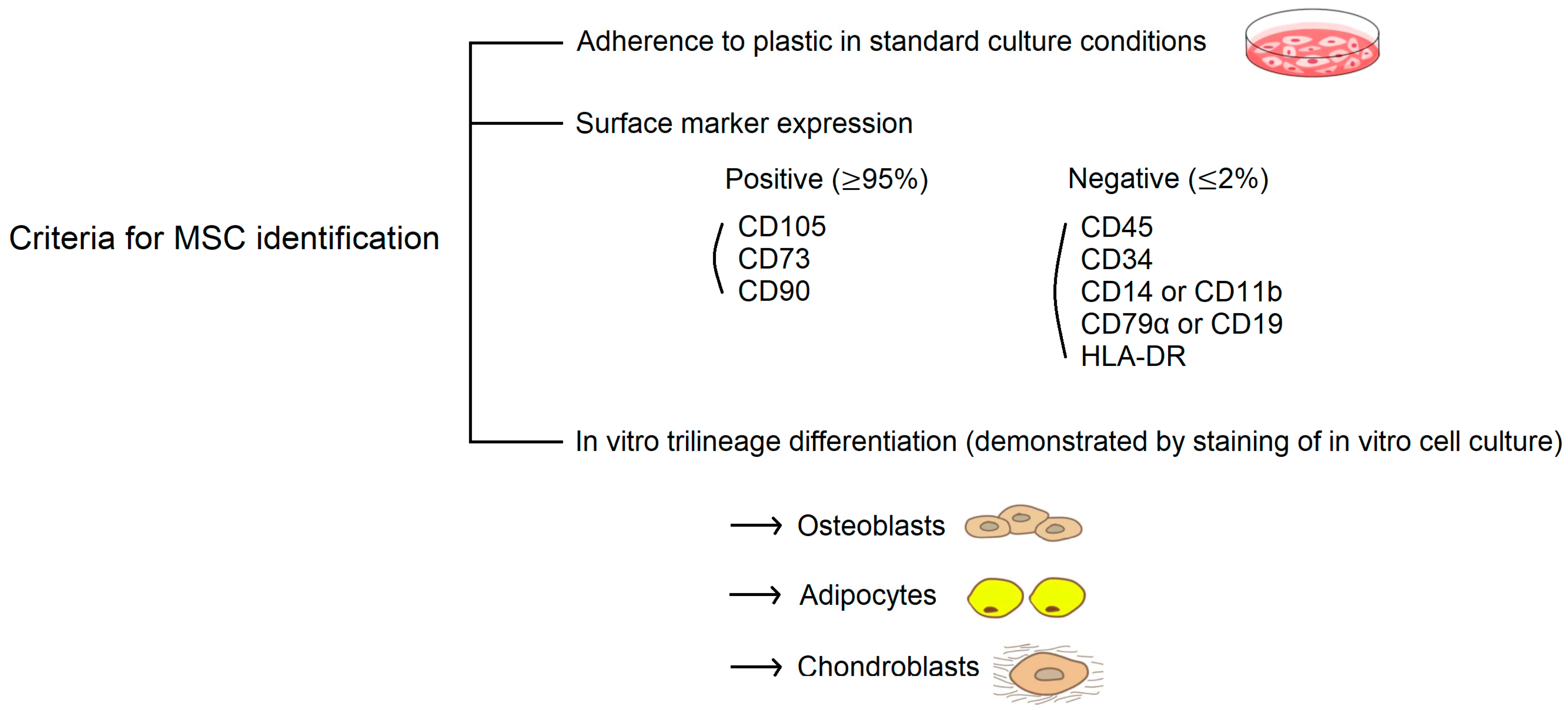
3.2. Properties of MSCs
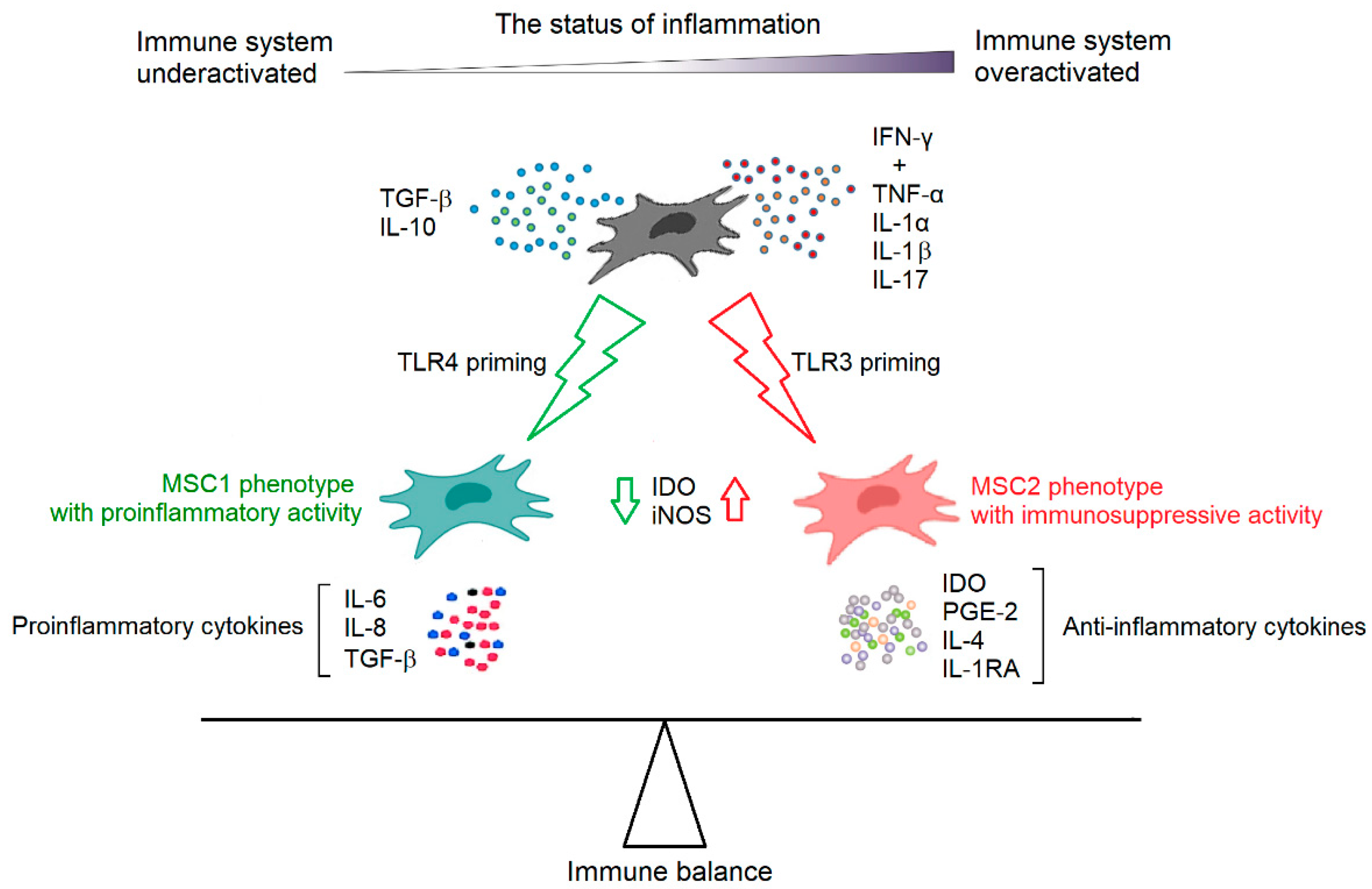
3.3. Immunomodulation by MSCs
3.4. MSCs from Various Origins
4. Alterations to MSCs in AA
4.1. Hematopoietic Support
4.2. Proliferative Potential
4.3. Surface Marker Expression
4.4. Differentiation Capacity
4.5. Immunomodulation
4.6. Gene Expression
5. MSC Therapy in Animal Models of AA
6. Clinical Application of MSCs in AA
6.1. MSC Therapy in Human Diseases
6.2. Current Treatment for Patients with AA
6.2.1. MSC Infusion
6.2.2. MSC and HSC Co-Transplantation
6.3. Challenges and Road Ahead
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ehrlich, P. Ueber einem Fall von Anämie mit Bemerkungen über regenerative Veränderungen des Knochenmarks. Charite’-Annalen 1888, 13, 300–309. [Google Scholar]
- Guinan, E.C. Diagnosis and management of aplastic anemia. Hematology 2011, 2011, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.S.; Kaufman, D.W. The epidemiology of acquired aplastic anemia. Haematologica 2008, 93, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Liu, C.; Fu, R.; Wang, H.; Wang, J.; Liu, X.; Feng, L.; Li, L.; Liu, H.; Wang, H.; et al. CD8+HLA-DR+ T cells are increased in patients with severe aplastic anemia. Mol. Med. Rep. 2014, 10, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Nakao, S.; Takami, A.; Takamatsu, H.; Zeng, W.; Sugimori, N.; Yamazaki, H.; Miura, Y.; Ueda, M.; Shiobara, S.; Yoshioka, T.; et al. Isolation of a T-cell clone showing HLA-DRB1 0405-restricted cytotoxicity for hematopoietic cells in a patient with aplastic anemia. Blood 1997, 89, 3691–3699. [Google Scholar] [CrossRef] [Green Version]
- Maciejewski, J.; Selleri, C.; Anderson, S.; Young, N.S. Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood 1995, 85, 3183–3190. [Google Scholar] [CrossRef]
- Kordasti, S.; Marsh, J.; Al-Khan, S.; Jiang, J.; Smith, A.; Mohamedali, A.; Abellan, P.P.; Veen, C.; Costantini, B.; Kulasekararaj, A.G.; et al. Functional characterization of CD4+ T cells in aplastic anemia. Blood 2012, 1, 2033–2043. [Google Scholar] [CrossRef]
- Solomou, E.E.; Rezvani, K.; Mielke, S.; Malide, D.; Keyvanfar, K.; Visconte, V.; Kajigaya, S.; Barrett, A.J.; Young, N.S. Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood 2007, 110, 1603–1606. [Google Scholar] [CrossRef]
- de Latour, R.P.; Visconte, V.; Takaku, T.; Wu, C.; Erie, A.J.; Sarcon, A.K.; Desierto, M.J.; Scheinberg, P.; Keyvanfar, K.; Nunez, O.; et al. Th17 immune responses contribute to the pathophysiology of aplastic anemia. Blood 2010, 116, 4175–4184. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhang, J.; Shi, J.; Ge, M.; Li, X.; Shao, Y.; Yao, J.; Zheng, Y. Increased bone marrow (BM) plasma level of soluble CD30 and correlations with BM plasma level of interferon (IFN)-gamma, CD4/CD8 T-cell ratio and disease severity in aplastic anemia. PLoS ONE 2014, 9, e110787. [Google Scholar] [CrossRef]
- Sloand, E.; Kim, S.; Maciejewski, J.P.; Tisdale, J.; Follmann, D.; Young, N.S. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood 2002, 100, 1185–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, S.; Shukla, P.; Nityanand, S. Expression of interferon-gamma and tumor necrosis factor-alpha in bone marrow T cells and their levels in bone marrow plasma in patients with aplastic anemia. Ann. Hematol. 2005, 84, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.C.; Chang, J.; Testa, N.G.; Hows, J.M.; Dexter, T.M. The hematopoietic defect in aplastic anemia assessed by long-term marrow culture. Blood 1990, 76, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manz, C.Y.; Nissen, C.; Wodnar-Filipowicz, A. Deficiency of CD34+ c-kit+ and CD34+38- hematopoietic precursors in aplastic anemia after immunosuppressive treatment. Am. J. Hematol. 1996, 52, 264–274. [Google Scholar] [CrossRef]
- Maciejewski, J.P.; Selleri, C.; Sato, T.; Anderson, S.; Young, N.S. A severe and consistent deficit in marrow and circulating primitive hematopoietic cells (long-term culture-initiating cells) in acquired aplastic anemia. Blood 1996, 88, 1983–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, S.; Scopes, J.; Elebute, M.O.; Papadaki, H.A.; Gordon-Smith, E.C.; Gibson, F.M. Stem cell defect in aplastic anemia: Reduced long term culture-initiating cells (LTC-IC) in CD34+ cells isolated from aplastic anemia patient bone marrow. Hematol. J. 2002, 3, 230–236. [Google Scholar] [CrossRef]
- Timeus, F.; Crescenzio, N.; Doria, A.; Foglia, L.; Linari, A.; Giaccone, M.; Pastore, G.; di Montezemolo, L.C.; Ramenghi, U.; Saracco, P. Flow cytometric evaluation of circulating CD34+ cell counts and apoptotic rate in children with acquired aplastic anemia and myelodysplasia. Exp. Hematol. 2005, 33, 597–604. [Google Scholar] [CrossRef]
- Callera, F.; Falcao, R.P. Increased apoptotic cells in bone marrow biopsies from patients with aplastic anaemia. Br. J. Haematol. 1997, 98, 18–20. [Google Scholar] [CrossRef]
- Maciejewski, J.P.; Selleri, C.; Sato, T.; Anderson, S.; Young, N.S. Increased expression of Fas antigen on bone marrow CD34+ cells of patients with aplastic anaemia. Br. J. Haematol. 1995, 91, 245–252. [Google Scholar] [CrossRef]
- Ismail, M.; Gibson, F.M.; Gordon-Smith, E.C.; Rutherford, T.R. Bcl-2 and Bcl-x expression in the CD34+ cells of aplastic anaemia patients: Relationship with increased apoptosis and upregulation of Fas antigen. Br. J. Haematol. 2001, 113, 706–712. [Google Scholar] [CrossRef]
- Zeng, W.; Chen, G.; Kajigaya, S.; Nunez, O.; Charrow, A.; Billings, E.M.; Young, N.S. Gene expression profiling in CD34 cells to identify differences between aplastic anemia patients and healthy volunteers. Blood 2004, 103, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Brummendorf, T.H.; Maciejewski, J.P.; Mak, J.; Young, N.S.; Lansdorp, P.M. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood 2001, 97, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, P.; Cooper, J.N.; Sloand, E.M.; Wu, C.O.; Calado, R.T.; Young, N.S. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA 2010, 304, 1358–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, H.; Nishio, N.; Hama, A.; Kawashima, N.; Wang, X.; Narita, A.; Doisaki, S.; Xu, Y.; Muramatsu, H.; Yoshida, N.; et al. Peripheral blood lymphocyte telomere length as a predictor of response to immunosuppressive therapy in childhood aplastic anemia. Haematologica 2014, 99, 1312–1316. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Calado, R.T.; Ly, H.; Kajigaya, S.; Baerlocher, G.M.; Chanock, S.J.; Lansdorp, P.M.; Young, N.S. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 2005, 352, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Baerlocher, G.M.; Lansdorp, P.M.; Chanock, S.J.; Nunez, O.; Sloand, E.; Young, N.S. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood 2003, 102, 916–918. [Google Scholar] [CrossRef] [Green Version]
- Akram, Z.; Ahmed, P.; Kajigaya, S.; Satti, T.M.; Satti, H.S.; Chaudhary, Q.U.N.; Gutierrez-Rodrigues, F.; Ibanez, P.F.; Feng, X.; Mahmood, S.K.; et al. Epidemiological, clinical and genetic characterization of aplastic anemia patients in Pakistan. Ann. Hematol. 2019, 98, 301–312. [Google Scholar] [CrossRef]
- Shao, W.; Tian, D.; Liu, C.; Sun, X.; Zhang, X. Aplastic anemia is associated with HLA-DRB1 1501 in northern Han Chinese. Int. J. Hematol. 2000, 71, 350–352. [Google Scholar]
- Babushok, D.V.; Perdigones, N.; Perin, J.C.; Olson, T.S.; Ye, W.; Roth, J.J.; Lind, C.; Cattier, C.; Li, Y.; Hartung, H.; et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015, 208, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Yoshizato, T.; Dumitriu, B.; Hosokawa, K.; Makishima, H.; Yoshida, K.; Townsley, D.; Sato-Otsubo, A.; Sato, Y.; Liu, D.; Suzuki, H.; et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N. Engl. J. Med. 2015, 1, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Kulasekararaj, A.G.; Jiang, J.; Smith, A.E.; Mohamedali, A.M.; Mian, S.; Gandhi, S.; Gaken, J.; Czepulkowski, B.; Marsh, J.C.; Mufti, G.J. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood 2014, 124, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Mo, W.; Zhang, Y.; Deng, H.; Li, Y.; Zhou, R.; Zhang, L.; Pan, S.; Wang, S. Impairment of hematopoietic stem cell niches in patients with aplastic anemia. Int. J. Hematol. 2015, 102, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Deans, R.J.; Moseley, A.B. Mesenchymal stem cells: Biology and potential clinical uses. Exp. Hematol. 2000, 28, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Owen, M. Lineage of osteogenic cells and their relationship to the stromal system. Bone Miner. Res. 1985, 3, 25. [Google Scholar]
- Wexler, S.A.; Donaldson, C.; Denning-Kendall, P.; Rice, C.; Bradley, B.; Hows, J.M. Adult bone marrow is a rich source of human mesenchymal ‘stem’ cells but umbilical cord and mobilized adult blood are not. Br. J. Haematol. 2003, 121, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Lazennec, G.; Jorgensen, C. Concise review: Adult multipotent stromal cells and cancer: Risk or benefit? Stem Cells 2008, 26, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- De Ugarte, D.A.; Alfonso, Z.; Zuk, P.A.; Elbarbary, A.; Zhu, M.; Ashjian, P.; Benhaim, P.; Hedrick, M.H.; Fraser, J.K. Differential expression of stem cell mobilization-associated molecules on multi-lineage cells from adipose tissue and bone marrow. Immunol. Lett. 2003, 89, 267–270. [Google Scholar] [CrossRef]
- Conget, P.A.; Minguell, J.J. Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J. Cell. Physiol. 1999, 181, 67–73. [Google Scholar] [CrossRef]
- da Silva, C.L.; Goncalves, R.; Crapnell, K.B.; Cabral, J.M.S.; Zanjani, E.D.; Almeida-Porada, G. A human stromal-based serum-free culture system supports the ex vivo expansion/maintenance of bone marrow and cord blood hematopoietic stem/progenitor cells. Exp. Hematol. 2005, 33, 828–835. [Google Scholar] [CrossRef]
- Van Overstraeten-Schlogel, N.; Beguin, Y.; Gothot, A. Role of stromal-derived factor-1 in the hematopoietic-supporting activity of human mesenchymal stem cells. Eur. J. Haematol. 2006, 76, 488–493. [Google Scholar] [CrossRef]
- Li, N.; Feugier, P.; Serrurrier, B.; Latger-Cannard, V.; Lesesve, J.F.; Stoltz, J.F.; Eljaafari, A. Human mesenchymal stem cells improve ex vivo expansion of adult human CD34+ peripheral blood progenitor cells and decrease their allostimulatory capacity. Exp. Hematol. 2007, 35, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Carrancio, S.; Blanco, B.; Romo, C.; Muntion, S.; Lopez-Holgado, N.; Blanco, J.F.; Brinon, J.G.; San Miguel, J.F.; Sanchez-Guijo, F.M.; del Canizo, M.C. Bone marrow mesenchymal stem cells for improving hematopoietic function: An in vitro and in vivo model. Part 2: Effect on bone marrow microenvironment. PLoS ONE 2011, 6, e26241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida-Porada, G.; Porada, C.D.; Tran, N.; Zanjani, E.D. Cotransplantation of human stromal cell progenitors into preimmune fetal sheep results in early appearance of human donor cells in circulation and boosts cell levels in bone marrow at later time points after transplantation. Blood 2000, 95, 3620–3627. [Google Scholar] [CrossRef] [PubMed]
- Noort, W.A.; Kruisselbrink, A.B.; Anker, P.S.I.; Kruger, M.; van Bezooijen, R.L.; de Paus, R.A.; Heemskerk, M.H.M.; Lowik, C.W.G.M.; Falkenburg, J.H.; Willemze, R.; et al. Mesenchymal stem cells promote engraftment of human umbilical cord blood-derived CD34(+) cells in NOD/SCID mice. Exp. Hematol. 2002, 30, 870–878. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Potian, J.A.; Aviv, H.; Ponzio, N.M.; Harrison, J.S.; Rameshwar, P. Veto-like activity of mesenchymal stem cells: Functional discrimination between cellular responses to alloantigens and recall antigens. J. Immunol. 2003, 171, 3426–3434. [Google Scholar] [CrossRef] [Green Version]
- Klyushnenkova, E.; Mosca, J.D.; Zernetkina, V.; Majumdar, M.K.; Beggs, K.J.; Simonetti, D.W.; Deans, R.J.; McIntosh, K.R. T cell responses to allogeneic human mesenchymal stem cells: Immunogenicity, tolerance, and suppression. J. Biomed. Sci. 2005, 12, 47–57. [Google Scholar] [CrossRef]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.J.; McTaggart, S.J. Immunosuppression by mesenchymal stromal cells: From culture to clinic. Exp. Hematol. 2008, 36, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J.; Sensebe, L. Mesenchymal stromal cells: Clinical challenges and therapeutic opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Pelagiadis, I.; Dimitriou, H.; Kalmanti, M. Biologic characteristics of mesenchymal stromal cells and their clinical applications in pediatric patients. J. Pediatr. Hematol./Oncol. 2008, 30, 301–309. [Google Scholar] [CrossRef]
- Krampera, M.; Cosmi, L.; Angeli, R.; Pasini, A.; Liotta, F.; Andreini, A.; Santarlasci, V.; Mazzinghi, B.; Pizzolo, G.; Vinante, F.; et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006, 24, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Su, J.; Zhang, L.; Zhao, X.; Ling, W.; L’Huillie, A.; Zhang, J.; Lu, Y.; Roberts, A.I.; Ji, W.; et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells 2009, 27, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- de Witte, S.F.H.; Merino, A.M.; Franquesa, M.; Strini, T.; van Zoggel, J.A.A.; Korevaar, S.S.; Luk, F.; Gargesha, M.; O’Flynn, L.; Roy, D.; et al. Cytokine treatment optimises the immunotherapeutic effects of umbilical cord-derived MSC for treatment of inflammatory liver disease. Stem Cell Res. Ther. 2017, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Yang, Q.; Lin, L.; Xu, C.; Zheng, C.; Chen, X.; Han, Y.; Li, M.; Cao, W.; Cao, K.; et al. Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ. 2014, 21, 1758–1768. [Google Scholar] [CrossRef]
- English, K.; Barry, F.P.; Field-Corbett, C.P.; Mahon, B.P. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol. Lett. 2007, 110, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yu, P.; Han, X.; Du, L.; Gan, J.; Wang, Y.; Shi, Y. TGF-beta promotes immune responses in the presence of mesenchymal stem cells. J. Immunol. 2014, 192, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, X.; Cao, W.; Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Djouad, F.; Toupet, K.; Bony, C.; Franquesa, M.; Hoogduijn, M.J.; Jorgensen, C.; Noel, D. Mesenchymal stem cell-derived interleukin 1 receptor antagonist promotes macrophage polarization and inhibits B cell differentiation. Stem Cells 2016, 34, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melief, S.M.; Schrama, E.; Brugman, M.H.; Tiemessen, M.M.; Hoogduijn, M.J.; Fibbe, W.E.; Roelofs, H. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells 2013, 31, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Fazekasova, H.; Lam, E.W.F.; Soeiro, I.; Lombardi, G.; Dazzi, F. Mesenchymal stem cells inhibit dendritic cell differentiation and function by preventing entry into the cell cycle. Transplantation 2007, 83, 71–76. [Google Scholar] [CrossRef]
- Du Rocher, B.; Mencalha, A.L.; Gomes, B.E.; Abdelhay, E. Mesenchymal stromal cells impair the differentiation of CD14(++) CD16(-) CD64(+) classical monocytes into CD14(++) CD16(+) CD64(++) activate monocytes. Cytotherapy 2012, 14, 12–25. [Google Scholar] [CrossRef]
- Chen, P.; Huang, Y.; Womer, K.L. Effects of mesenchymal stromal cells on human myeloid dendritic cell differentiation and maturation in a humanized mouse model. J. Immunol. Methods 2015, 427, 100–104. [Google Scholar] [CrossRef]
- Jiang, X.X.; Zhang, Y.; Liu, B.; Zhang, S.X.; Wu, Y.; Yu, X.D.; Mao, N. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood 2005, 105, 4120–4126. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Ge, W.; Li, C.; You, S.; Liao, L.; Han, Q.; Deng, W.; Zhao, R.C. Effects of mesenchymal stem cells on differentiation, maturation, and function of human monocyte-derived dendritic cells. Stem Cells Dev. 2004, 13, 263–271. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Y.; Ye, L.; Zhang, T.; Cheng, J.; Chen, G.; Zhang, Q.; Yang, Y. Umbilical cord-derived mesenchymal stem cells instruct monocytes towards an IL10-producing phenotype by secreting IL6 and HGF. Sci. Rep. 2016, 6, 37566. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.D.; Kosaka, Y.; Marcus, P.; Rashedi, I.; Keating, A. Differential immunomodulatory effects of human bone marrow-derived mesenchymal stromal cells on natural killer cells. Stem Cells Dev. 2019, 28, 933–943. [Google Scholar] [CrossRef]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate B-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Franquesa, M.; Mensah, F.K.; Huizinga, R.; Strini, T.; Boon, L.; Lombardo, E.; DelaRosa, O.; Laman, J.D.; Grinyó, J.M.; Weimar, W.; et al. Human adipose tissue-derived mesenchymal stem cells abrogate plasmablast formation and induce regulatory B cells independently of T helper cells. Stem Cells 2015, 33, 880–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asari, S.; Itakura, S.; Ferreri, K.; Liu, C.P.; Kuroda, Y.; Kandeel, F.; Mullen, Y. Mesenchymal stem cells suppress B-cell terminal differentiation. Exp. Hematol. 2009, 37, 604–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafei, M.; Hsieh, J.; Fortier, S.; Li, M.; Yuan, S.; Birman, E.; Forner, K.; Boivin, M.N.; Doody, K.; Tremblay, M.; et al. Mesenchymal stromal cell-derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood 2008, 112, 4991–4998. [Google Scholar] [CrossRef]
- Carter, N.A.; Vasconcellos, R.; Rosser, E.C.; Tulone, C.; Munoz-Suano, A.; Kamanaka, M.; Ehrenstein, M.R.; Flavell, R.A.; Mauri, C. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J. Immunol. 2011, 186, 5569–5579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, L.C.; Heldring, N.; Kadri, N.; Le Blanc, K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells 2017, 35, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, B.; Wang, D.; Ji, Y.; Kong, Q.; Wang, G.; Wang, J.; Zhao, W.; Jin, L.; Li, H. Murine bone marrow mesenchymal stem cells cause mature dendritic cells to promote T-cell tolerance. Scand. J. Immunol. 2008, 68, 607–615. [Google Scholar] [CrossRef]
- Marson, A.; Kretschmer, K.; Frampton, G.M.; Jacobsen, E.S.; Polansky, J.K.; MacIsaac, K.D.; Levine, S.S.; Fraenkel, E.; von Boehmer, H.; Young, R.A. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 2007, 445, 931–935. [Google Scholar] [CrossRef] [Green Version]
- Ge, W.; Jiang, J.; Arp, J.; Liu, W.; Garcia, B.; Wang, H. Regulatory T-cell generation and kidney allograft tolerance induced by mesenchymal stem cells associated with indoleamine 2,3-dioxygenase expression. Transplantation 2010, 90, 1312–1320. [Google Scholar] [CrossRef]
- English, K.; Ryan, J.M.; Tobin, L.; Murphy, M.J.; Barry, F.P.; Mahon, B.P. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin. Exp. Immunol. 2009, 156, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Rashedi, I.; Gómez-Aristizábal, A.; Wang, X.H.; Viswanathan, S.; Keating, A. TLR3 or TLR4 activation enhances mesenchymal stromal cell-mediated Treg induction via Notch signaling. Stem Cells 2017, 35, 265–275. [Google Scholar] [CrossRef]
- Ghannam, S.; Pene, J.; Torcy-Moquet, G.; Jorgensen, C.; Yssel, H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J. Immunol. 2010, 185, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Li, B. The functional stability of FOXP3 and RORγt in Treg and Th17 and their therapeutic applications. Adv. Protein Chem. Struct. Biol. 2017, 107, 155–189. [Google Scholar] [CrossRef]
- Terraza-Aguirre, C.; Campos-Mora, M.; Elizondo-Vega, R.; Contreras-Lopez, R.A.; Luz-Crawford, P.; Jorgensen, C.; Djouad, F. Mechanisms behind the immunoregulatory dialogue between mesenchymal stem cells and Th17 cells. Cells 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Piatetzky-Shapiro, I.I.; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. Development 1966, 16, 381–390. [Google Scholar] [CrossRef]
- Secco, M.; Zucconi, E.; Vieira, N.M.; Fogaca, L.L.; Cerqueira, A.; Carvalho, M.D.; Jazedje, T.; Okamoto, O.K.; Muotri, A.R.; Zatz, M. Multipotent stem cells from umbilical cord: Cord is richer than blood! Stem Cells 2008, 26, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Guillot, P.V.; Gotherstrom, C.; Chan, J.; Kurata, H.; Fisk, N.M. Human first-trimester fetal MSC express pluripotency markers and grow faster and have longer telomeres than adult MSC. Stem Cells 2007, 25, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.; Eichler, H.; Stoeve, J.; Kluter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef]
- Baksh, D.; Yao, R.; Tuan, R.S. Comparison of proliferative and multilineage differentiation potential of human mesenchymal stem cells derived from umbilical cord and bone marrow. Stem Cells 2007, 25, 1384–1392. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Teoh, S.H.; Chong, M.S.K.; Schantz, J.T.; Fisk, N.M.; Choolani, M.A.; Chan, J. Superior osteogenic capacity for bone tissue engineering of fetal compared with perinatal and adult mesenchymal stem cells. Stem Cells 2009, 27, 126–137. [Google Scholar] [CrossRef]
- Gotherstrom, C.; Ringden, O.; Westgren, M.; Tammik, C.; Le Blanc, K. Immunomodulatory effects of human foetal liver-derived mesenchymal stem cells. Bone Marrow Transpl. 2003, 32, 265–272. [Google Scholar] [CrossRef]
- Chan, C.K.; Wu, K.H.; Lee, Y.S.; Hwang, S.M.; Lee, M.S.; Liao, S.K.; Cheng, E.H.; See, L.C.; Tsai, C.N.; Kuo, M.L.; et al. The comparison of interleukin 6-associated immunosuppressive effects of human ESCs, fetal-type MSCs, and adult-type MSCs. Transplantation. Transplantation 2012, 94, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Hotta, T.; Kato, T.; Maeda, H.; Yamao, H.; Yamada, H.; Saito, H. Functional changes in marrow stromal cells in aplastic anaemia. Acta Haematol. 1985, 74, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, L.A.; Seidel, K.; Leisenring, W.; Torok-Storb, B. Aplastic anemia: Analysis of stromal cell function in long-term marrow cultures. Blood 1994, 84, 3685–3690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.H.; Lin, C.W.; Pan, H.H.; Yang, S.F.; Weng, T.F.; Peng, C.T.; Wu, K.H. Increased apoptosis and peripheral blood mononuclear cell suppression of bone marrow mesenchymal stem cells in severe aplastic anemia. Pediatr. Blood Cancer 2018, 65, e27247. [Google Scholar] [CrossRef] [PubMed]
- Hamzic, E.; Whiting, K.; Gordon Smith, E.; Pettengell, R. Characterization of bone marrow mesenchymal stromal cells in aplastic anaemia. Br. J. Haematol. 2015, 169, 804–813. [Google Scholar] [CrossRef]
- Lu, S.; Ge, M.; Zheng, Y.; Li, J.; Feng, X.; Feng, S.; Huang, J.; Feng, Y.; Yang, D.; Shi, J.; et al. CD106 is a novel mediator of bone marrow mesenchymal stem cells via NF-kappaB in the bone marrow failure of acquired aplastic anemia. Stem Cell Res. Ther. 2017, 8, 178. [Google Scholar] [CrossRef]
- Chao, Y.H.; Peng, C.T.; Harn, H.J.; Chan, C.K.; Wu, K.H. Poor potential of proliferation and differentiation in bone marrow mesenchymal stem cells derived from children with severe aplastic anemia. Ann. Hematol. 2010, 89, 715–723. [Google Scholar] [CrossRef]
- El-Mahgoub, E.R.; Ahmed, E.; Afifi, R.A.E.A.; Kamal, M.A.; Mousa, S.M. Mesenchymal stem cells from pediatric patients with aplastic anemia: Isolation, characterization, adipogenic, and osteogenic differentiation. Fetal Pediatr. Pathol. 2014, 33, 9–15. [Google Scholar] [CrossRef]
- Huo, J.; Zhang, L.; Ren, X.; Li, C.; Li, X.; Dong, P.; Zheng, X.; Huang, J.; Shao, Y.; Ge, M.; et al. Multifaceted characterization of the signatures and efficacy of mesenchymal stem/stromal cells in acquired aplastic anemia. Stem Cell Res. Ther. 2020, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Qin, M.; Wu, R.; Meng, H.; He, Y.; Wang, B.; Zhou, X.; Zhu, G. Insensitive to PTH of CD8+ T cells regulate bone marrow mesenchymal stromal cell in aplastic anemia patients. Int. J. Med. Sci. 2020, 17, 1665–1672. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Xie, X.T.; Jiang, H.; Zhou, J.J.; Li, F.X.; Cao, P. Low expression of basic fibroblastic growth factor in mesenchymal stem cells and bone marrow of children with aplastic anemia. Pediatr. Hematol. Oncol. 2014, 31, 11–19. [Google Scholar] [CrossRef]
- Li, J.; Yang, S.; Lu, S.; Zhao, H.; Feng, J.; Li, W.; Ma, F.; Ren, Q.; Liu, B.; Zhang, L.; et al. Differential gene expression profile associated with the abnormality of bone marrow mesenchymal stem cells in aplastic anemia. PLoS ONE 2012, 7, e47764. [Google Scholar] [CrossRef] [Green Version]
- Bueno, C.; Roldan, M.; Anguita, E.; Romero-Moya, D.; Martin-Antonio, B.; Rosu-Myles, M.; del Canizo, C.; Campos, F.; Garcia, R.; Gomez-Casares, M.; et al. Bone marrow mesenchymal stem cells from patients with aplastic anemia maintain functional and immune properties and do not contribute to the pathogenesis of the disease. Haematologica 2014, 99, 1168–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atmar, K.; Tulling, A.J.; Lankester, A.C.; Bartels, M.; Smiers, F.J.; van der Burg, M.; Mohseny, A.B. Functional and immune modulatory characteristics of bone marrow mesenchymal stromal cells in patients with aplastic anemia: A systematic review. Front. Immunol. 2022, 13, 859668. [Google Scholar] [CrossRef] [PubMed]
- Bacigalupo, A.; Valle, M.; Podesta, M.; Pitto, A.; Zocchi, E.; De Flora, A.; Pozzi, S.; Luchetti, S.; Frassoni, F.; Van Lint, M.T.; et al. T-cell suppression mediated by mesenchymal stem cells is deficient in patients with severe aplastic anemia. Exp. Hematol. 2005, 33, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, S.; Yang, S.; Xing, W.; Feng, J.; Li, W.; Zhao, Q.; Wu, H.; Ge, M.; Ma, F.; et al. Impaired immunomodulatory ability of bone marrow mesenchymal stem cells on CD4(+) T cells in aplastic anemia. Results Immunol. 2012, 2, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Li, J.P.; Wu, K.H.; Chao, W.R.; Lee, Y.J.; Yang, S.F.; Chao, Y.H. Alterations of mesenchymal stem cells on regulating Th17 and Treg differentiation in severe aplastic anemia. Aging 2023, 15, 30. [Google Scholar] [CrossRef]
- Chao, Y.H.; Wu, K.H.; Chiou, S.H.; Chiang, S.F.; Huang, C.Y.; Yang, H.C.; Chan, C.K.; Peng, C.T.; Wu, H.P.; Chow, K.C.; et al. Downregulated CXCL12 expression in mesenchymal stem cells associated with severe aplastic anemia in children. Ann. Hematol. 2015, 94, 13–22. [Google Scholar] [CrossRef]
- Shipounova, I.N.; Petrova, T.V.; Svinareva, D.A.; Momotuk, K.S.; Mikhailova, E.A.; Drize, N.I.; Shipounova, I.N.; Petrova, T.V.; Svinareva, D.A.; Momotuk, K.S.; et al. Alterations in hematopoietic microenvironment in patients with aplastic anemia. Clin. Transl. Sci. 2009, 2, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.X.; Sun, Q.Y.; Guo, M.; Ai, H.S. The radiation protection and therapy effects of mesenchymal stem cells in mice with acute radiation injury. Br. J. Radiol. 2010, 83, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, S.; Lee, S.B.; Lee, J.G.; Jang, W.S.; Lee, S.J.; Park, S.; Lee, S.S. Mitigating effects of hUCB-MSCs on the hematopoietic syndrome resulting from total body irradiation. Exp. Hematol. 2013, 41, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Meng, F.; Zhou, X.; Li, C.; He, Y.; Zeng, X.; Jiang, X.; Liu, J.; Zeng, G.; Tang, Y.; et al. Hematopoietic recovery of acute radiation syndrome by human superoxide dismutase-expressing umbilical cord mesenchymal stromal cells. Cytotherapy 2015, 17, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.; Yanez, R.M.; Sanchez-Dominguez, R.; Hernando-Rodriguez, M.; Peces-Barba, M.; Herrera, G.; O’Connor, J.E.; Segovia, J.C.; Bueren, J.A.; Lamana, M.L. Mesenchymal stromal cells enhance the engraftment of hematopoietic stem cells in an autologous mouse transplantation model. Stem Cell Res. Ther. 2015, 6, 165. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, H.; Lv, Y.B.; Wang, Q.; Xie, Z.J.; Ma, L.H.; He, J.; Xue, W.; Yu, S.; Guo, J.; et al. Intraperitoneal injection of multiplacentas pooled cells treatment on a mouse model with aplastic anemia. Stem Cells Int. 2016, 2016, 3279793. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, S.; Zhou, Y.; Feng, F.; Wang, Q.; Zhu, X.; Zhao, J.; Fu, H.; Lv, M.; Ai, H.; et al. Adipose-derived mesenchymal stem cells (ADSCs) with the potential to ameliorate platelet recovery, enhance megakaryopoiesis, and inhibit apoptosis of bone marrow cells in a mouse model of radiation-induced thrombocytopenia. Cell Transpl. 2016, 25, 261–273. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Huang, F.; Wang, J.; Su, W.; Zhou, J.; Qi, Q.; Cao, F.; Sun, B.; Liu, Z.; et al. Human gingiva tissue-derived MSC ameliorates immune-mediated bone marrow failure of aplastic anemia via suppression of Th1 and Th17 cells and enhancement of CD4+Foxp3+ regulatory T cells differentiation. Am. J. Transl. Res. 2019, 11, 7627–7643. [Google Scholar]
- Galderisi, U.; Peluso, G.; Di Bernardo, G. Clinical trials based on mesenchymal stromal cells are exponentially increasing: Where are we in recent years? Stem Cell Rev. Rep. 2022, 18, 23–36. [Google Scholar] [CrossRef]
- Jovic, D.; Yu, Y.; Wang, D.; Wang, K.; Li, H.; Xu, F.; Liu, C.; Liu, J.; Luo, Y. A brief overview of global trends in MSC-based cell therapy. Stem Cell Rev. Rep. 2022, 18, 1525–1545. [Google Scholar] [CrossRef]
- Peslak, S.A.; Olson, T.; Babushok, D.V. Diagnosis and treatment of aplastic anemia. Curr. Treat. Options Oncol. 2017, 18, 70. [Google Scholar] [CrossRef]
- DeZern, A.E.; Guinan, E.C. Aplastic anemia in adolescents and young adults. Acta Haematol. 2014, 132, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Boddu, P.C.; Kadia, T.M. Updates on the pathophysiology and treatment of aplastic anemia: A comprehensive review. Expert Rev. Hematol. 2017, 10, 433–448. [Google Scholar] [CrossRef]
- Chao, Y.H.; Wu, H.P.; Chan, C.K.; Tsai, C.; Peng, C.T.; Wu, K.H. Umbilical cord-derived mesenchymal stem cells for hematopoietic stem cell transplantation. J. Biomed. Biotechnol. 2012, 2012, 759503. [Google Scholar] [CrossRef] [Green Version]
- Fouillard, L.; Bensidhoum, M.; Bories, D.; Bonte, H.; Lopez, M.; Moseley, A.M.; Smith, A.; Lesage, S.; Beaujean, F.; Thierry, D.; et al. Engraftment of allogeneic mesenchymal stem cells in the bone marrow of a patient with severe idiopathic aplastic anemia improves stroma. Leukemia 2003, 17, 474–476. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Jiang, Z.J.; Pang, Y.; Li, L.; Gao, Y.; Xiao, H.W.; Li, Y.H.; Zhang, H.; Liu, Q. Efficacy and safety of mesenchymal stromal cell treatment from related donors for patients with refractory aplastic anemia. Cytotherapy 2013, 15, 760–766. [Google Scholar] [CrossRef]
- Cle, D.V.; Santana-Lemos, B.; Tellechea, M.F.; Prata, K.L.; Orellana, M.D.; Covas, D.T.; Calado, R.T. Intravenous infusion of allogeneic mesenchymal stromal cells in refractory or relapsed aplastic anemia. Cytotherapy 2015, 17, 1696–1705. [Google Scholar] [CrossRef]
- Pang, Y.; Xiao, H.W.; Zhang, H.; Liu, Z.H.; Li, L.; Gao, Y.; Li, H.B.; Jiang, Z.J.; Tan, H.; Lin, J.R.; et al. Allogeneic bone marrow-derived mesenchymal stromal cells expanded in vitro for treatment of aplastic anemia: A multicenter phase II trial. Stem cells Transl. Med. 2017, 6, 1569–1575. [Google Scholar] [CrossRef]
- Lan, Y.; Liu, F.; Chang, L.; Liu, L.; Zhang, Y.; Yi, M.; Cai, Y.; Feng, J.; Han, Z.; Han, Z.; et al. Combination of umbilical cord mesenchymal stem cells and standard immunosuppressive regimen for pediatric patients with severe aplastic anemia. BMC Pediatr. 2021, 21, 102. [Google Scholar] [CrossRef]
- Jaganathan, B.G.; Tisato, V.; Vulliamy, T.; Dokal, I.; Marsh, J.; Dazzi, F.; Bonnet, D. Effects of MSC co-injection on the reconstitution of aplastic anemia patient following hematopoietic stem cell transplantation. Leukemia 2010, 24, 1791–1795. [Google Scholar] [CrossRef]
- Chao, Y.H.; Tsai, C.; Peng, C.T.; Wu, H.P.; Chan, C.K.; Weng, T.; Wu, K.H. Cotransplantation of umbilical cord MSCs to enhance engraftment of hematopoietic stem cells in patients with severe aplastic anemia. Bone Marrow Transpl. 2011, 46, 1391–1392. [Google Scholar] [CrossRef]
- Wang, H.; Yan, H.; Wang, Z.; Zhu, L.; Liu, J.; Guo, Z. Cotransplantation of allogeneic mesenchymal and hematopoietic stem cells in children with aplastic anemia. Pediatrics 2012, 129, e1612–e1615. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Yang, K.; Qin, M.; Zhang, C.; Du, Z.; Zhang, X.; Liu, Y.; Yue, Y.; Feng, Z. Efficacy and safety of human umbilical cord derived mesenchymal stem cell therapy in children with severe aplastic anemia following allogeneic hematopoietic stem cell transplantation: A retrospective case series of 37 patients. Pediatr. Hematol. Oncol. 2014, 31, 39–49. [Google Scholar] [CrossRef]
- Li, X.H.; Gao, C.J.; Da, W.M.; Cao, Y.B.; Wang, Z.H.; Xu, L.X.; Wu, Y.M.; Liu, B.; Liu, Z.Y.; Yan, B.; et al. Reduced intensity conditioning, combined transplantation of haploidentical hematopoietic stem cells and mesenchymal stem cells in patients with severe aplastic anemia. PLoS ONE 2014, 9, e89666. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, Y.; Xiao, H.; Yao, Z.; Zhang, H.; Liu, Q.; Wu, B.; Nie, D.; Li, Y.; Pang, Y.; et al. Cotransplantation of bone marrow-derived mesenchymal stem cells in haploidentical hematopoietic stem cell transplantation in patients with severe aplastic anemia: An interim summary for a multicenter phase II trial results. Bone Marrow Transpl. 2017, 52, 704–710. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Z.; Wu, Y.; Yang, X.; Cao, Y.; Li, X.; Yan, B.; Li, S.; Da, W.; Wu, X. Clinical evaluation of haploidentical hematopoietic combined with human umbilical cord-derived mesenchymal stem cells in severe aplastic anemia. Eur. J. Med. Res. 2018, 23, 12. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yu, H.; Cao, F.; Liu, Z.; Liu, Z.; Feng, W.; Liu, X.; Yu, Y.; Xiao, Y.; Li, L.; et al. Donor-derived marrow mesenchymal stromal cell co-transplantation following a haploidentical hematopoietic stem cell transplantation trail to treat severe aplastic anemia in children. Ann. Hematol. 2019, 98, 473–479. [Google Scholar] [CrossRef]
- Ding, L.; Han, D.M.; Zheng, X.L.; Yan, H.M.; Xue, M.; Liu, J.; Zhu, L.; Guo, Z.K.; Mao, N.; Ning, H.M.; et al. Infusion of haploidentical hematopoietic stem cells combined with mesenchymal stem cells for treatment of severe aplastic anemia in adult patients yields curative effects. Cytotherapy 2022, 24, 205–212. [Google Scholar] [CrossRef]
- Sheng, X.F.; Li, H.; Hong, L.L.; Zhuang, H. Combination of haploidentical hematopoietic stem cell transplantation with umbilical cord-derived mesenchymal stem cells in patients with severe aplastic anemia: A retrospective controlled study. Turk. J. Hematol. 2022, 39, 117–129. [Google Scholar] [CrossRef]
- Troyer, D.L.; Weiss, M.L. Wharton’s jelly-derived cells are a primitive stromal cell population. Stem Cells 2008, 26, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Gotherstrom, C.; West, A.; Liden, J.; Uzunel, M.; Lahesmaa, R.; Le Blanc, K. Difference in gene expression between human fetal liver and adult bone marrow mesenchymal stem cells. Haematologica 2005, 90, 1017–1026. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Animals | Attempt to Induce AA | MSC Source | MSC Administration | Efficacy of MSC Therapy |
|---|---|---|---|---|---|
| [111] | BALB/c mice | IR (5.5 Gy) | Allogenic BM | 2.5 × 107/kg, IV, once, 4 h after IR | Rapid recovery of blood cells; lower apoptotic ratio of BM cells; increase in BM hematopoietic islands; recovery of CFU-GM and CFU-F |
| [111] | BALB/c mice | IR (8 Gy) | Allogenic BM | Three groups: 2.5, 5, or 15 × 107/kg, IV, once, cotransplanted with 1 × 109/kg of donor BM cells | Improvements in survival, but not in those receiving 15 × 107/kg MSCs |
| [112] | BALB/c mice | IR (7 Gy) | Human cord blood | 2 × 106, IV, once, 4 h after IR | Greater proliferation of BM and PB cells; better hematopoietic reconstitution |
| [113] | BALB/c mice | IR (5.8 Gy) | Human umbilical cords + ECSOD | 1 × 106, IV, twice, 1 h and 48 h after IR | Improvements in survival; promoted hematopoietic recovery; decrease in radiation-induced O2- and apoptosis |
| [114] | B6D2F1 mice | IR (5 Gy) | Autologous adipose tissue | 6 × 105, IV, once, cotransplanted with HSCs | Improvements in hematopoietic reconstitution; enhanced donor HSC engraftment; facilitated migration and homing of donor HSCs |
| [115] | BALB/cBy mice | IR (4 Gy) + lymph node cell infusion | Allogenic multiplacenta pooled cells | 1 × 107/kg, IP, once, 7 days after IR | Longer survival time; improved PB hemoglobin levels, but not BM architecture response |
| [116] | BALB/c mice | IR (4 Gy) | Rat adipose tissue | 2 × 106, IV, once, immediately after IR | Better recovery of platelets and leukocytes in PB, but not RBCs; increase in BM total CFUs and megakaryocyte-CFUs; improved BM cellularity; inhibited apoptosis of BM cells; antiapoptotic effects mediated via the PI3K/Akt pathway |
| [117] | CB6F1 mice | IR (5 Gy) + lymph node cell infusion | Human gingiva tissue | 2 × 106, IV, once, 6 days after IR | Improvements in survival; attenuated T cells-mediated BM damage; protective effects by regulating the balance of Th1, Th17, and Tregs |
| [100] | CByB6F1 mice | IR (5 Gy) + lymph node cell infusion | Human umbilical cords | 1 × 106, IV, once, 3 days after IR | Improvements in survival; alleviate body weight decline; ameliorated pathological damages; regained hematopoiesis and immunoregulatory capacity |
| Reference | Number of Patients | Disease Status (SAA/NSAA) | MSC Source | MSC Administration | Main Findings |
|---|---|---|---|---|---|
| [124] | 1 | Refractory SAA | Allogeneic relative BM | Twice (2 × 106/kg and 6 × 106/kg), IV | No effects on hematopoiesis; partial recovery of BM stroma; MSC engraftment detected in recipient BM; death due to invasive fungal infection |
| [125] | 18 | Refractory AA (4/14) | Allogeneic relative BM | 5.0–7.1 × 105/kg, IV, once | Achieved CR or PR at 1 year in 6 patients; no major adverse events; increase in Tregs in PB |
| [126] | 9 | Refractory or relapsed AA (7/2) | Allogeneic HLA- mismatched unrelative BM | 1.3–4.5 × 106/kg, IV, weekly, 2–5 times | Achieved PR at 6 months in only 2 patients; no significant improvement in clinical hematologic responses; no infusion-related adverse events; four deaths due to heart failure and infections; no evidence for MSC engraftment in recipient BM |
| [127] | 74 | Refractory AA (24/50) | Allogeneic BM | 1–2 × 106/kg, IV, weekly, for 4 weeks | 28.4% overall response (6.8% CR; 21.6% PR); 87.8% overall survival at 2 years; no significant adverse events |
| [128] | 9 | Newly diagnosed SAA in children | Allogeneic umbilical cords | 1 × 106/kg, IV, weekly, for 3 weeks | Compared to that in patients receiving IST alone, no significant differences in early response rates and long-term outcomes |
| Reference | Patient Number | Status | MSC Source | MSC Administration | Main Findings |
|---|---|---|---|---|---|
| [129] | 1 | Refractory SAA for MUD PBSCT | Third-party donor | 1 × 106/kg, twice, 1st dose on day 0 and 2nd boost on day + 26 | Hematopoietic engraftment; sustained remission |
| [130] | 2 | Children with refractory SAA for MUD PBSCT | 5/6 MUD umbilical cords | 4.2–4.3 × 106/kg, once, 4 h before HSC infusion | Faster neutrophil and platelet engraftment; no acute or chronic GVHD; no infusion-related adverse events |
| [131] | 6 | Children with refractory SAA for HSCT | Umbilical cords or BM | 0.85–2.5 × 106/kg, once, before HSC infusion | Stable HSC engraftment; no severe acute GVHD; no infusion-related adverse events |
| [132] | 37 | Pediatric patients with refractory SAA for allogeneic HSCT | Umbilical cords | 0.78–3.41 × 106/kg, one (n = 20) or more times (n = 17), infusion frequency depending on GVHD severity | Evidence of proliferative BM; 100% successful HSC engraftment; 74.2% OS; decrease in death due to infection and organ failure; no effective control in patients with severe GVHD and concomitant serious infection; no significant adverse effects |
| [133] | 17 | Refractory SAA for haploidentical HSCT | Umbilical cords | 2.87–10 × 106/kg, 6 h before HSC infusion | No primary graft failure; 23.5% grade III–IV acute GVHD; 14.2% moderate and severe chronic GVHD; 76.5% OS at 6 months; no infusion-related adverse events |
| [134] | 44 | SAA for haploidentical HSCT | Allogeneic BM | 3.2–4.1 × 106/kg, 1st dose on day 0 and 2nd dose on day + 14, additional doses weekly for 1–4 weeks if poor graft function and severe GVHD | Reduced graft failure and severe GVHD in haploidentical HSCT: 97.6% hematopoietic reconstitution and full donor chimerism, 29.3% grade II–IV acute GVHD, 14.6% chronic GVHD; 77.3% OS; no infusion toxicity |
| [135] | 24 | SAA for haploidentical HSCT | Umbilical cords | 5 × 105/kg, once, 4 h before HSC infusion | All achieved donor chimerism within 1 month; 50% acute GVHD; 83.3% OS at 6 months |
| [136] | 33 | Children with SAA for haploidentical HSCT | Donor BM | 1 × 106/kg, twice, 1st dose on day 0 (6 h before HSC infusion) and 2nd dose on day + 14 | Faster hematopoietic implantation; 100% hematopoietic reconstitution and full donor chimerism; effective prevention of severe GVHD (25.71% grade II–IV acute GVHD, 22.86% chronic GVHD); 85.71% OS; no infusion toxicity |
| [137] | 25 | SAA for haploidentical HSCT | Umbilical cords | 1 × 106/kg, once, on day 0 | All achieved neutrophil engraftment; achieved platelet engraftment in 23 patients; 32% grade II acute GVHD; no grade III–IV acute GVHD; 28% chronic GVHD; 71.78% OS |
| [138] | 47 | SAA for haploidentical HSCT | Umbilical cords | 1 × 106/kg, once, 4 h before HSC infusion | Compared to those without MSC infusion, faster neutrophil engraftment, lower cumulative incidence of chronic GVHD, similar rate of acute GVHD, better 5-year OS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.-A.; Li, J.-P.; Wu, K.-H.; Yang, S.-F.; Chao, Y.-H. Mesenchymal Stem Cells in Acquired Aplastic Anemia: The Spectrum from Basic to Clinical Utility. Int. J. Mol. Sci. 2023, 24, 4464. https://doi.org/10.3390/ijms24054464
Wang X-A, Li J-P, Wu K-H, Yang S-F, Chao Y-H. Mesenchymal Stem Cells in Acquired Aplastic Anemia: The Spectrum from Basic to Clinical Utility. International Journal of Molecular Sciences. 2023; 24(5):4464. https://doi.org/10.3390/ijms24054464
Chicago/Turabian StyleWang, Xing-An, Ju-Pi Li, Kang-Hsi Wu, Shun-Fa Yang, and Yu-Hua Chao. 2023. "Mesenchymal Stem Cells in Acquired Aplastic Anemia: The Spectrum from Basic to Clinical Utility" International Journal of Molecular Sciences 24, no. 5: 4464. https://doi.org/10.3390/ijms24054464
APA StyleWang, X. -A., Li, J. -P., Wu, K. -H., Yang, S. -F., & Chao, Y. -H. (2023). Mesenchymal Stem Cells in Acquired Aplastic Anemia: The Spectrum from Basic to Clinical Utility. International Journal of Molecular Sciences, 24(5), 4464. https://doi.org/10.3390/ijms24054464