
Dissecting the Meiotic Recombination Patterns in a Brassica napus Double Haploid Population Using 60K SNP Array

 ,
, 
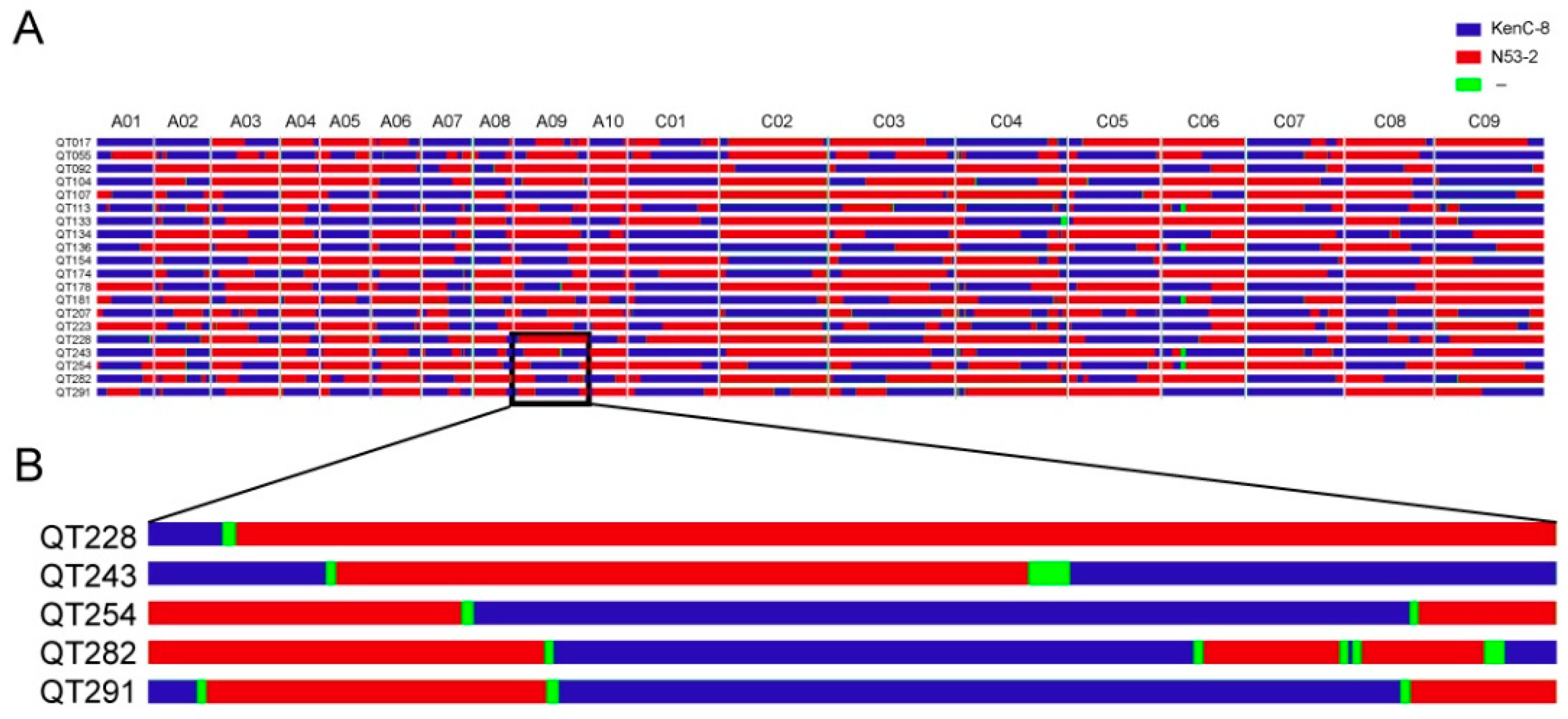{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genotyping the DH Population in B. napus using the Brassica 60K Array
2.2. Bin Detection and CO Distribution
2.3. CO Hot Regions and Their Genes
2.4. Homologous Genome Exchanges May Occur between the Subgenomes
2.5. Bin Map Construction and Plant Phenotype Study
3. Discussion
4. Materials and Methods
4.1. Plant Materials and DNA Extraction
4.2. Genotyping and SNP Identification
4.3. Bin and CO Validation
4.4. Gene Analysis of the CO Hot Regions
4.5. Bin Map Construction and Plant Phenotype Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, K.; Higgins, J.D.; Sanchez Moran, E.; Armstrong, S.J.; Franklin, F.C.H. Pathways to meiotic recombination in Arabidopsis thaliana. New Phytol. 2011, 190, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.; Cohen, P.E. Control of Meiotic Crossovers: From Double-Strand Break Formation to Designation. Annu. Rev. Genet. 2016, 50, 175–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.; Liu, X.; Hu, L.; Song, Q.; Liu, S.; Huang, Q.; Geng, Z.; Zhu, Y.; Li, X.; Fu, F.; et al. VE-822, a novel DNA Holliday junction stabilizer, inhibits homologous recombination repair and triggers DNA damage response in osteogenic sarcomas. Biochem. Pharmacol. 2021, 193, 114767. [Google Scholar] [CrossRef]
- Rommel Fuentes, R.; Hesselink, T.; Nieuwenhuis, R.; Bakker, L.; Schijlen, E.; Dooijeweert, W.; Diaz Trivino, S.; Haan, J.R.; Sanchez Perez, G.; Zhang, X.; et al. Meiotic recombination profiling of interspecific hybrid F1 tomato pollen by linked read sequencing. Plant J. 2020, 102, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Borner, G.V.; Kleckner, N.; Hunter, N. Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 2004, 117, 29–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manhart, C.M.; Alani, E. Roles for mismatch repair family proteins in promoting meiotic crossing over. DNA Repair 2016, 38, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Mercier, R.; Mézard, C.; Jenczewski, E.; Macaisne, N.; Grelon, M. The Molecular Biology of Meiosis in Plants. Annu. Rev. Plant Biol. 2015, 66, 297–327. [Google Scholar] [CrossRef]
- Wang, C.; Higgins, J.D.; He, Y.; Lu, P.; Zhang, D.; Liang, W. Resolvase OsGEN1 Mediates DNA Repair by Homologous Recombination. Plant Physiol. 2017, 173, 1316–1329. [Google Scholar] [CrossRef]
- Bhérer, C.; Campbell, C.L.; Auton, A. Refined genetic maps reveal sexual dimorphism in human meiotic recombination at multiple scales. Nat. Commun. 2017, 8, 14994. [Google Scholar] [CrossRef] [Green Version]
- Giraut, L.; Falque, M.; Drouaud, J.; Pereira, L.; Martin, O.C.; Mezard, C. Genome-wide crossover distribution in Arabidopsis thaliana meiosis reveals sex-specific patterns along chromosomes. PLoS Genet. 2011, 7, e1002354. [Google Scholar] [CrossRef] [Green Version]
- Argueso, J.L.; Wanat, J.; Gemici, Z.; Alani, E. Competing Crossover Pathways Act During Meiosis in Saccharomyces cerevisiae. Genetics 2004, 168, 1805–1816. [Google Scholar] [CrossRef] [Green Version]
- Hunter, N.; Kleckner, N. The single-end invasion: An asymmetric intermediate at the double-strand break to double-holliday junction transition of meiotic recombination. Cell 2001, 106, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishant, K.T.; Plys, A.J.; Alani, E. A Mutation in the Putative MLH3 Endonuclease Domain Confers a Defect in Both Mismatch Repair and Meiosis in Saccharomyces cerevisiae. Genetics 2008, 179, 747–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iovene, M.; Wielgus, S.M.; Simon, P.W.; Buell, C.R.; Jiang, J. Chromatin Structure and Physical Mapping of Chromosome 6 of Potato and Comparative Analyses with Tomato. Genetics 2008, 180, 1307–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhu, X.; Zhang, M.; Chao, S.; Xu, S.; Cai, X. Meiotic homoeologous recombination-based mapping of wheat chromosome 2B and its homoeologues in Aegilops speltoides and Thinopyrum elongatum. Theor. Appl. Genet. 2018, 131, 2381–2395. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Szinay, D.; Lang, C.; Ramanna, M.S.; van der Vossen, E.A.G.; Datema, E.; Lankhorst, R.K.; de Boer, J.; Peters, S.A.; Bachem, C.; et al. Cross-Species Bacterial Artificial Chromosome–Fluorescencein Situ Hybridization Painting of the Tomato and Potato Chromosome 6 Reveals Undescribed Chromosomal Rearrangements. Genetics 2008, 180, 1319–1328. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, L.; Yan, J. Dissecting meiotic recombination based on tetrad analysis by single-microspore sequencing in maize. Nat. Commun. 2015, 6, 6648. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Li, X.; Zhang, Q.; Yan, J. Single gametophyte sequencing reveals that crossover events differ between sexes in maize. Nat. Commun. 2019, 10, 785. [Google Scholar] [CrossRef] [Green Version]
- Kianian, P.M.A.; Wang, M.; Simons, K.; Ghavami, F.; He, Y.; Dukowic-Schulze, S.; Sundararajan, A.; Sun, Q.; Pillardy, J.; Mudge, J.; et al. High-resolution crossover mapping reveals similarities and differences of male and female recombination in maize. Nat. Commun. 2018, 9, 2370. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.J.; Fuchs, J.; Houben, A.; Heckmann, S. High-throughput measuring of meiotic recombination rates in barley pollen nuclei using Crystal Digital PCRTM. Plant J. 2021, 107, 649–661. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Inaba, R.; Nishio, T. Phylogenetic analysis of Brassiceae based on the nucleotide sequences of the S-locus related gene, SLR1. Theor. Appl. Genet. 2002, 105, 1159–1165. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Long, Y.; Li, D.; Yin, Y.; Tian, J.; Chen, L.; Liu, L.; Zhao, W.; Zhao, Y.; et al. Identification of QTLs associated with oil content in a high-oil Brassica napus cultivar and construction of a high-density consensus map for QTLs comparison in B. napus. PLoS ONE 2013, 8, e80569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Tong, C.; Zhang, X.; Song, A.; Hu, M.; Dong, W.; Chen, F.; Wang, Y.; Tu, J.; Liu, S.; et al. A high-quality Brassica napus genome reveals expansion of transposable elements, subgenome evolution and disease resistance. Plant Biotechnol. J. 2021, 19, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Li, H.; Chao, H.; He, J.; Ding, Y.; Zhao, W.; Zhang, K.; Xiong, Y.; Chen, K.; Zhang, L.; et al. Refinement of four major QTL for oil content in Brassica napus by integration of genome resequencing and transcriptomics. Crop J. 2022, 10, 627–637. [Google Scholar] [CrossRef]
- Chao, H.; Wang, H.; Wang, X.; Guo, L.; Gu, J.; Zhao, W.; Li, B.; Chen, D.; Raboanatahiry, N.; Li, M. Genetic dissection of seed oil and protein content and identification of networks associated with oil content in Brassica napus. Sci. Rep. 2017, 7, 46295. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, G.K.; Fang, C.; Olson, M.A.; Falque, M.; Martin, O.C.; Pawlowski, W.P. Recombination patterns in maize reveal limits to crossover homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 15982–15987. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, S.D.; Ogle, D.E.; Ayoub, M.A.; Heckmann, S.; Henderson, I.R.; Edwards, K.J.; Higgins, J.D. MutS homologue 4 and MutS homologue 5 Maintain the Obligate Crossover in Wheat Despite Stepwise Gene Loss following Polyploidization. Plant Physiol. 2020, 183, 1545–1558. [Google Scholar] [CrossRef]
- Gruhn, J.R.; Rubio, C.; Broman, K.W.; Hunt, P.A.; Hassold, T. Cytological studies of human meiosis: Sex-specific differences in recombination originate at, or prior to, establishment of double-strand breaks. PLoS ONE 2013, 8, e85075. [Google Scholar] [CrossRef] [Green Version]
- Froenicke, L.; Anderson, L.K.; Wienberg, J.; Ashley, T. Male mouse recombination maps for each autosome identified by chromosome painting. Am. J. Hum. Genet. 2002, 71, 1353–1368. [Google Scholar] [CrossRef] [Green Version]
- Wijnker, E.; Velikkakam James, G.; Ding, J.; Becker, F.; Klasen, J.R.; Rawat, V.; Rowan, B.A.; de Jong, D.F.; de Snoo, C.B.; Zapata, L.; et al. The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. eLife 2013, 2, e01426. [Google Scholar] [CrossRef]
- Lynn, A.; Koehler, K.E.; Judis, L.; Chan, E.R.; Cherry, J.P.; Schwartz, S.; Seftel, A.; Hunt, P.A.; Hassold, T.J. Covariation of synaptonemal complex length and mammalian meiotic exchange rates. Science 2002, 296, 2222–2225. [Google Scholar] [CrossRef]
- Wang, S.; Hassold, T.; Hunt, P.; White, M.A.; Zickler, D.; Kleckner, N.; Zhang, L. Inefficient Crossover Maturation Underlies Elevated Aneuploidy in Human Female Meiosis. Cell 2017, 168, 977–989. [Google Scholar] [CrossRef] [Green Version]
- Kong, A.; Thorleifsson, G.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Walters, G.B.; Jonasdottir, A.; Gylfason, A.; Kristinsson, K.T.; et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 2010, 467, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhao, Z.; Zheng, X.; Zhou, J.; Kong, W.; Wang, P.; Bai, W.; Zheng, H.; Zhang, H.; Li, J.; et al. A selfish genetic element confers non-Mendelian inheritance in rice. Science 2018, 360, 1130–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawe, R.K.; Lowry, E.G.; Gent, J.I.; Stitzer, M.C.; Swentowsky, K.W.; Higgins, D.M.; Ross-Ibarra, J.; Wallace, J.G.; Kanizay, L.B.; Alabady, M.; et al. A Kinesin-14 Motor Activates Neocentromeres to Promote Meiotic Drive in Maize. Cell 2018, 173, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Francis, K.E.; Lam, S.Y.; Harrison, B.D.; Bey, A.L.; Berchowitz, L.E.; Copenhaver, G.P. Pollen tetrad-based visual assay for meiotic recombination in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 3913–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, A. Crossover patterning in plants. Plant Reprod. 2022. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D. Salicylic acid signaling in disease resistance. Plant Sci. 2014, 228, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Bhatia, G.; Garewal, N.; Upadhyay, A.; Singh, K. Identification of defense related gene families and their response against powdery and downy mildew infections in Vitis vinifera. BMC Genom. 2021, 22, 776. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, E.; Falque, M.; Walter, H.; Bauland, C.; Camisan, C.; Campo, L.; Meyer, N.; Ranc, N.; Rincent, R.; Schipprack, W.; et al. Intraspecific variation of recombination rate in maize. Genome Biol. 2013, 14, R103. [Google Scholar] [CrossRef]
- Blary, A.; Jenczewski, E. Manipulation of crossover frequency and distribution for plant breeding. Theor. Appl. Genet. 2019, 132, 575–592. [Google Scholar] [CrossRef] [Green Version]
- Pelé, A.; Falque, M.; Trotoux, G.; Eber, F.; Nègre, S.; Gilet, M.; Huteau, V.; Lodé, M.; Jousseaume, T.; Dechaumet, S.; et al. Amplifying recombination genome-wide and reshaping crossover landscapes in Brassicas. PLoS Genet. 2017, 13, e1006794. [Google Scholar] [CrossRef] [Green Version]
- Boideau, F.; Pelé, A.; Tanguy, C.; Trotoux, G.; Eber, F.; Maillet, L.; Gilet, M.; Lodé-Taburel, M.; Huteau, V.; Morice, J.; et al. A Modified Meiotic Recombination in Brassica napus Largely Improves Its Breeding Efficiency. Biology 2021, 10, 771. [Google Scholar] [CrossRef]
- Grandont, L.; Cuñado, N.; Coriton, O.; Huteau, V.; Eber, F.; Chèvre, A.M.; Grelon, M.; Chelysheva, L.; Jenczewski, E. Homoeologous Chromosome Sorting and Progression of Meiotic Recombination in Brassica napus: Ploidy Does Matter! Plant Cell 2014, 26, 1448–1463. [Google Scholar] [CrossRef] [Green Version]
- Higgins, E.E.; Clarke, W.E.; Howell, E.C.; Armstrong, S.J.; Parkin, I. Detecting de Novo Homoeologous Recombination Events in Cultivated Brassica napus Using a Genome-Wide SNP Array. G3 2018, 8, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Gaeta, R.T.; Edger, P.P.; Cao, Y.; Zhao, K.; Zhang, S.; Pires, J.C. Chromosome inheritance and meiotic stability in allopolyploid Brassica napus. G3 2021, 11, jkaa011. [Google Scholar] [CrossRef]
- He, Z.; Wang, L.; Harper, A.L.; Havlickova, L.; Pradhan, A.K.; Parkin, I.A.P.; Bancroft, I. Extensive homoeologous genome exchanges in allopolyploid crops revealed by mRNAseq-based visualization. Plant Biotechnol. J. 2017, 15, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Dalton-Morgan, J.; Hayward, A.; Alamery, S.; Tollenaere, R.; Mason, A.S.; Campbell, E.; Patel, D.; Lorenc, M.T.; Yi, B.; Long, Y.; et al. A high-throughput SNP array in the amphidiploid species Brassica napus shows diversity in resistance genes. Funct. Integr. Genom. 2014, 14, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Fan, L.; Zhang, Z.; Yang, X.; Liu, Y.; Ma, Y.; Pan, Y.; Zhou, G.; Zhang, M.; Ning, H.; et al. Global dissection of the recombination landscape in soybean using a high-density 600K SoySNP array. Plant Biotechnol. J. 2022. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Ooijen, J.W. JoinMap 4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Wageningen University & Research: Wageningen, The Netherlands, 2006; 59p. [Google Scholar]
- Wang, H.; Guo, J.; Lambert, K.N.; Lin, Y. Developmental control of Arabidopsis seed oil biosynthesis. Planta 2007, 226, 773–783. [Google Scholar] [CrossRef]
- Li, B.; Zhao, W.; Li, D.; Chao, H.; Zhao, X.; Ta, N.; Li, Y.; Guan, Z.; Guo, L.; Zhang, L.; et al. Genetic dissection of the mechanism of flowering time based on an environmentally stable and specific QTL in Brassica napus. Plant Sci. 2018, 277, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Li, H.; Yan, S.; Zhao, W.; Chen, K.; Wang, H.; Raboanatahiry, N.; Huang, J.; Li, M. Further insight into decreases in seed glucosinolate content based on QTL mapping and RNA-seq in Brassica napus L. Theor. Appl. Genet. 2022, 135, 2969–2991. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Chao, H.; Chen, L.; Wang, H.; Zhao, W.; Li, B.; Zhang, L.; Li, H.; Wang, B.; Li, M. Stable and novel QTL identification and new insights into the genetic networks affecting seed fiber traits in Brassica napus. Theor. Appl. Genet. 2019, 132, 1761–1775. [Google Scholar] [CrossRef]
- Zhao, W.; Chao, H.; Zhang, L.; Ta, N.; Zhao, Y.; Li, B.; Zhang, K.; Guan, Z.; Hou, D.; Chen, K.; et al. Integration of QTL Mapping and Gene Fishing Techniques to Dissect the Multi-Main Stem Trait in Rapeseed (Brassica napus L.). Front. Plant Sci. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Wang, X.; Wang, H.; Tian, J.; Li, B.; Chen, L.; Chao, H.; Long, Y.; Xiang, J.; Gan, J.; et al. Genome-Wide Identification of QTL for Seed Yield and Yield-Related Traits and Construction of a High-Density Consensus Map for QTL Comparison in Brassica napus. Front. Plant Sci. 2016, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; He, J.; Tang, M.; Ming, B.; Li, H.; Fan, S.; Xiong, Y.; Chao, H.; Zhang, L.; Wang, A.; et al. Dissecting the Meiotic Recombination Patterns in a Brassica napus Double Haploid Population Using 60K SNP Array. Int. J. Mol. Sci. 2023, 24, 4469. https://doi.org/10.3390/ijms24054469
Yan S, He J, Tang M, Ming B, Li H, Fan S, Xiong Y, Chao H, Zhang L, Wang A, et al. Dissecting the Meiotic Recombination Patterns in a Brassica napus Double Haploid Population Using 60K SNP Array. International Journal of Molecular Sciences. 2023; 24(5):4469. https://doi.org/10.3390/ijms24054469
Chicago/Turabian StyleYan, Shuxiang, Jianjie He, Mi Tang, Bangfa Ming, Huaixin Li, Shipeng Fan, Yiyi Xiong, Hongbo Chao, Libin Zhang, Aihua Wang, and et al. 2023. "Dissecting the Meiotic Recombination Patterns in a Brassica napus Double Haploid Population Using 60K SNP Array" International Journal of Molecular Sciences 24, no. 5: 4469. https://doi.org/10.3390/ijms24054469
APA StyleYan, S., He, J., Tang, M., Ming, B., Li, H., Fan, S., Xiong, Y., Chao, H., Zhang, L., Wang, A., & Li, M. (2023). Dissecting the Meiotic Recombination Patterns in a Brassica napus Double Haploid Population Using 60K SNP Array. International Journal of Molecular Sciences, 24(5), 4469. https://doi.org/10.3390/ijms24054469