
Genetic Adaptation of Siberian Larch (Larix sibirica Ledeb.) to High Altitudes



Abstract
:1. Introduction
2. Results
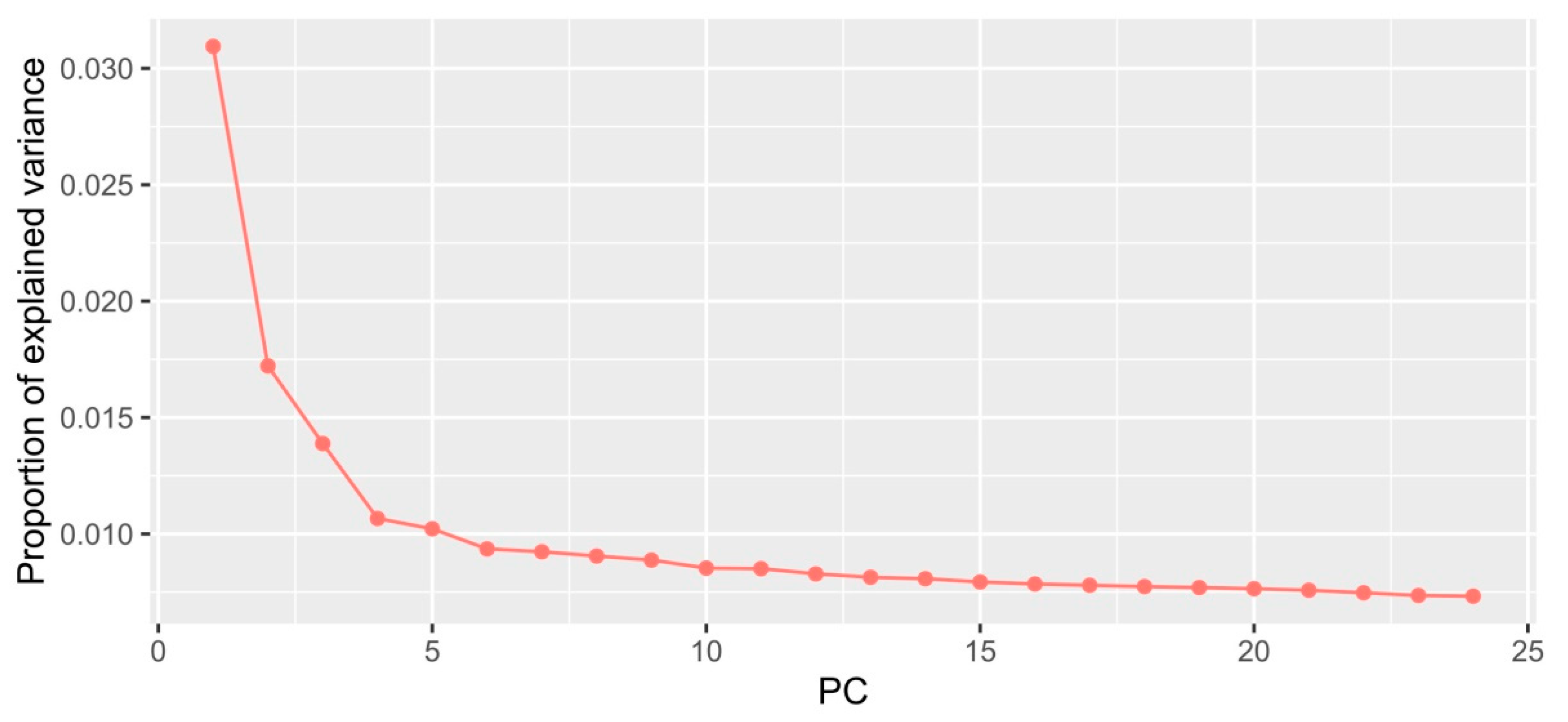
2.1. Environmental Variables
2.2. SNP Dataset
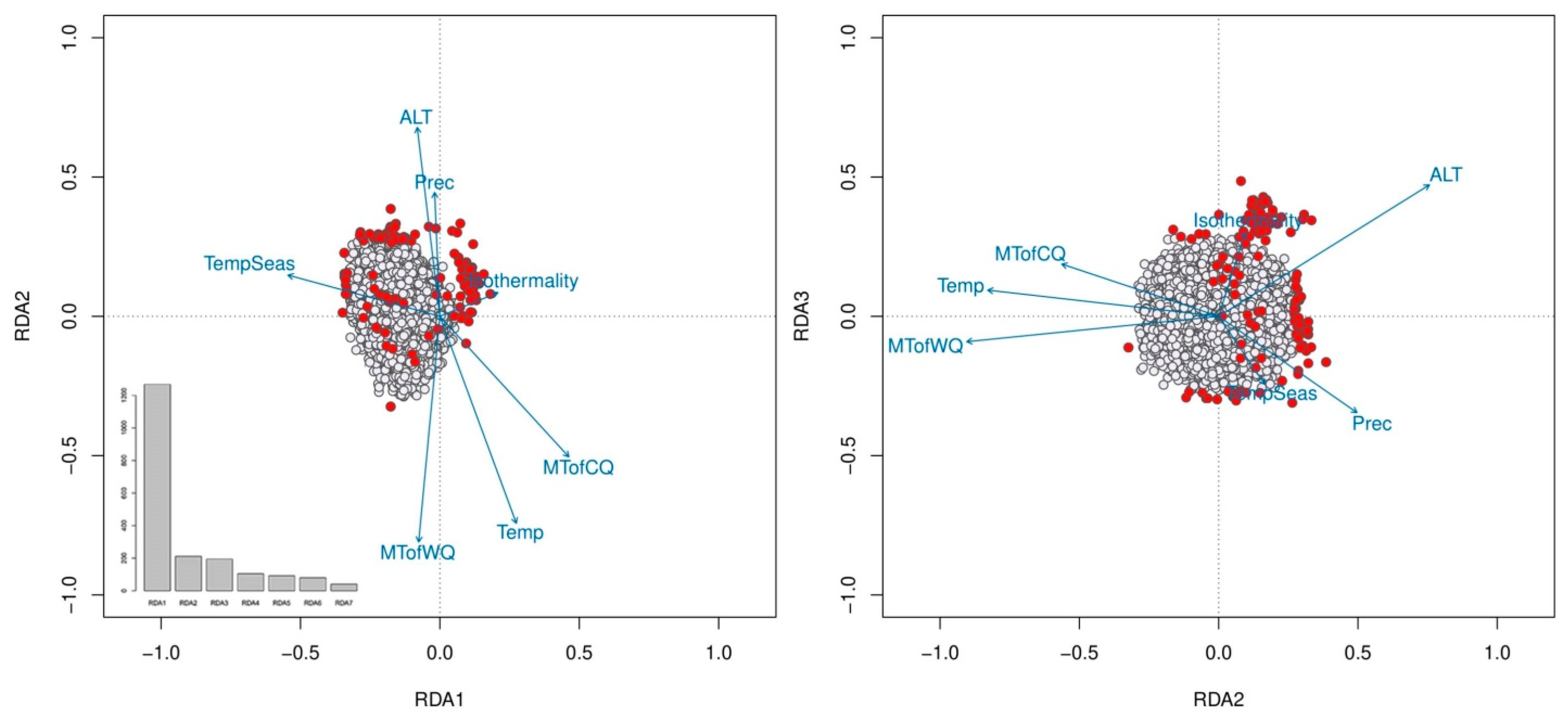
2.3. Detection of SNPs Associated with Environmental Variables and Outliers
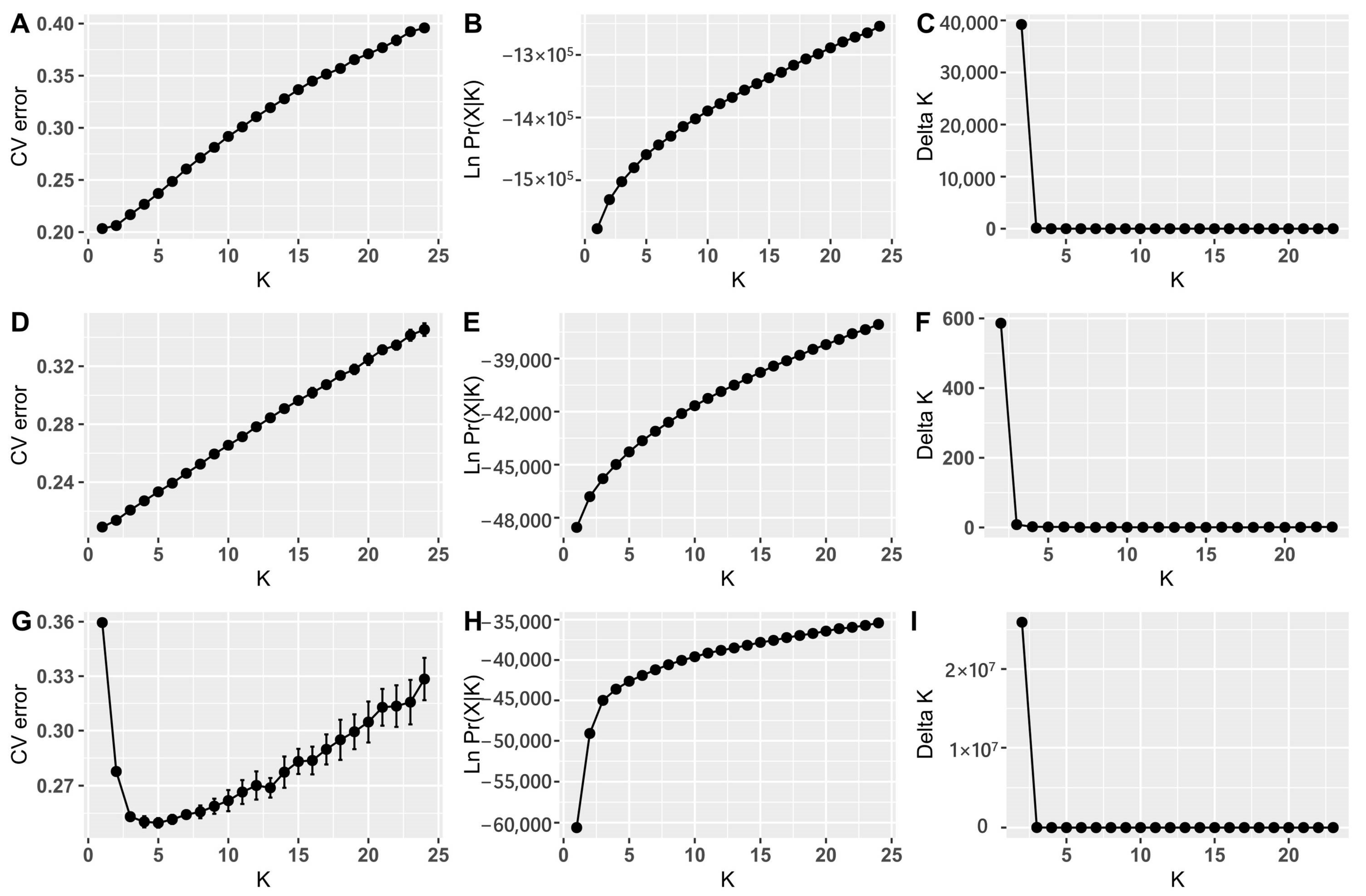
2.4. Population Genetic Variation, Structure and Differentiation
2.5. SNP Annotation
3. Discussion
4. Materials and Methods
4.1. Plant Material and DNA Isolation
4.2. Library Construction
4.3. Bioclimatic Data
4.4. SNP Calling
4.5. Detection of SNPs Associated with Environmental Variables and Outliers
4.6. Population Genetic Variation, Structure and Differentiation
4.7. SNP Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohri, D.; Khoshoo, T.N. Genome Size in Gymnosperms. Plant Syst. Evol. 1986, 153, 119–132. [Google Scholar] [CrossRef]
- Kuzmin, D.A.; Feranchuk, S.I.; Sharov, V.V.; Cybin, A.N.; Makolov, S.V.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Stepwise large genome assembly approach: A case of Siberian larch (Larix sibirica Ledeb). BMC Bioinform. 2019, 20 (Suppl. 1), 37. [Google Scholar] [CrossRef] [Green Version]
- Bondar, E.I.; Feranchuk, S.I.; Miroshnikova, K.A.; Sharov, V.V.; Kuzmin, D.A.; Oreshkova, N.V.; Krutovsky, K.V. Annotation of Siberian Larch (Larix sibirica Ledeb.) Nuclear Genome—One of the Most Cold-Resistant Tree Species in the Only Deciduous GENUS in Pinaceae. Plants 2022, 11, 2062. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Joost, S.; Vuilleumier, S.; Jensen, J.D.; Schoville, S.; Leempoel, K.; Stucki, S.; Widmer, I.; Melodelima, C.; Rolland, J.; Manel, S. Uncovering the genetic basis of adaptive change: On the intersection of landscape genomics and theoretical population genetics. Mol. Ecol. 2013, 22, 3659–3665. [Google Scholar] [CrossRef] [PubMed]
- Balkenhol, N.; Dudaniec, R.Y.; Krutovsky, K.V.; Johnson, J.S.; Cairns, D.M.; Segelbacher, G.; Selkoe, K.A.; von der Heyden, S.; Wang, I.J.; Selmoni, O.; et al. Landscape Genomics: Understanding Relationships Between Environmental Heterogeneity and Genomic Characteristics of Populations. In Population Genomics: Concepts, Approaches and Applications; Rajora, O.P., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 261–322. ISBN 978-3-030-04589-0. [Google Scholar]
- Haddad, N.M.; Brudvig, L.A.; Clobert, J.; Davies, K.F.; Gonzalez, A.; Holt, R.D.; Lovejoy, T.E.; Sexton, J.O.; Austin, M.P.; Collins, C.D.; et al. Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci. Adv. 2015, 1, e1500052. [Google Scholar] [CrossRef] [Green Version]
- Fahrig, L. Effects of Habitat Fragmentation on Biodiversity. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 487–515. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, K.R. Climate change and its impact on biodiversity and human welfare. Proc. Indian Natl. Sci. Acad. 2022, 88, 160–171. [Google Scholar] [CrossRef]
- Feng, L.; Du, F.K. Landscape Genomics in Tree Conservation Under a Changing Environment. Front. Plant Sci. 2022, 13, 822217. [Google Scholar] [CrossRef]
- Narum, S.R.; Hess, J.E. Comparison of FST outlier tests for SNP loci under selection: Analytical approaches. Mol. Ecol. Resour. 2011, 11, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.R.; Josephs, E.B.; Morris, G.P. Genotype–environment associations to reveal the molecular basis of environmental adaptation. Plant Cell 2022, 35, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F. Using genome scans of DNA polymorphism to infer adaptive population divergence: Genome scans and adaptive population divergence. Mol. Ecol. 2005, 14, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Coop, G.; Witonsky, D.; Di Rienzo, A.; Pritchard, J.K. Using Environmental Correlations to Identify Loci Underlying Local Adaptation. Genetics 2010, 185, 1411–1423. [Google Scholar] [CrossRef] [Green Version]
- Eckert, A.J.; Bower, A.D.; Wegrzyn, J.L.; Pande, B.; Jermstad, K.D.; Krutovsky, K.V.; St Clair, J.B.; Neale, D.B. Association Genetics of Coastal Douglas Fir (Pseudotsuga menziesii var. menziesii, Pinaceae). I. Cold-Hardiness Related Traits. Genetics 2009, 182, 1289–1302. [Google Scholar] [CrossRef] [Green Version]
- De Kort, H.; Vandepitte, K.; Bruun, H.H.; Closset-Kopp, D.; Honnay, O.; Mergeay, J. Landscape genomics and a common garden trial reveal adaptive differentiation to temperature across Europe in the tree species Alnus glutinosa. Mol. Ecol. 2014, 23, 4709–4721. [Google Scholar] [CrossRef]
- Zheng, C.; Tan, L.; Sang, M.; Ye, M.; Wu, R. Genetic adaptation of Tibetan poplar (Populus szechuanica var. tibetica) to high altitudes on the Qinghai-Tibetan Plateau. Ecol. Evol. 2020, 10, 10974–10985. [Google Scholar] [CrossRef]
- Privé, F.; Luu, K.; Vilhjálmsson, B.J.; Blum, M.G.B. Performing Highly Efficient Genome Scans for Local Adaptation with R Package pcadapt Version 4. Mol. Biol. Evol. 2020, 37, 2153–2154. [Google Scholar] [CrossRef]
- Caye, K.; Jumentier, B.; Lepeule, J.; François, O. LFMM 2: Fast and Accurate Inference of Gene-Environment Associations in Genome-Wide Studies. Mol. Biol. Evol. 2019, 36, 852–860. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Mussmann, S.M.; Douglas, M.R.; Chafin, T.K.; Douglas, M.E. AdmixPipe: Population Analyses in Admixture for Non-Model Organisms. BMC Bioinform. 2020, 21, 337. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Bagnoli, F.; Fady, B.; Fineschi, S.; Oddou-Muratorio, S.; Piotti, A.; Sebastiani, F.; Vendramin, G.G. Neutral patterns of genetic variation and applications to conservation in conifer species. In Genetics, Genomics and Breeding of Conifers, 1st ed.; Plomion, C., Bousquet, J., Kole, C., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 141–195. ISBN 9781578087198. [Google Scholar]
- Neale, D.B.; Wheeler, N.C. Neutral Genetic Variation. In The Conifers: Genomes, Variation and Evolution, 1st ed.; Springer Nature: Cham, Switzerland, 2019; pp. 181–224. [Google Scholar] [CrossRef]
- Krutovsky, K.; Burczyk, J.; Chybicki, I. Gene Flow, Spatial Structure, Local Adaptation, and Assisted Migration in Trees. In Genomics of Tree Crops, 1st ed.; Schnell, R.J., Priyadarshan, P.M., Eds.; Springer: New York, NY, USA; Berlin/Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2012; pp. 71–116. ISBN 9781461409199. [Google Scholar]
- Gao, J.; Lan, T. Functional Characterization of the Late Embryogenesis Abundant (LEA) Protein Gene Family from Pinus tabuliformis (Pinaceae) in Escherichia coli. Sci. Rep. 2016, 6, 19467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneeshaw, S.; Keyani, R.; Delorme-Hinoux, V.; Imrie, L.; Loake, G.J.; Le Bihan, T.; Reichheld, J.-P.; Spoel, S.H. Nucleoredoxin guards against oxidative stress by protecting antioxidant enzymes. Proc. Natl. Acad. Sci. USA 2017, 114, 8414–8419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipreos, E.T.; Pagano, M. The F-box protein family. Genome Biol. 2000, 1, reviews3002.1. [Google Scholar] [CrossRef] [Green Version]
- Schröder, F.; Lisso, J.; Lange, P.; Müssig, C. The extracellular EXO protein mediates cell expansion in Arabidopsis leaves. BMC Plant Biol. 2009, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klink, V.P.; Lawaju, B.R.; Niraula, P.M.; Sharma, K.; McNeece, B.T.; Pant, S.R.; Troell, H.A.; Acharya, S.; Khatri, R.; Rose, A.H.; et al. The conserved oligomeric Golgi (COG) complex, a window into plant-pathogen interactions. J. Plant Interact. 2022, 17, 344–360. [Google Scholar] [CrossRef]
- Domergue, F.; Vishwanath, S.J.; Joubès, J.; Ono, J.; Lee, J.A.; Bourdon, M.; Alhattab, R.; Lowe, C.; Pascal, S.; Lessire, R.; et al. Three Arabidopsis Fatty Acyl-Coenzyme A Reductases, FAR1, FAR4, and FAR5, Generate Primary Fatty Alcohols Associated with Suberin Deposition. Plant Physiol. 2010, 153, 1539–1554. [Google Scholar] [CrossRef] [Green Version]
- Di Rubbo, S.; Irani, N.G.; Kim, S.Y.; Xu, Z.-Y.; Gadeyne, A.; Dejonghe, W.; Vanhoutte, I.; Persiau, G.; Eeckhout, D.; Simon, S.; et al. The Clathrin Adaptor Complex AP-2 Mediates Endocytosis of BRASSINOSTEROID INSENSITIVE1 in Arabidopsis. Plant Cell 2013, 25, 2986–2997. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cnops, G.; Vanderhaeghen, R.; De Block, S.; Van Montagu, M.; Van Lijsebettens, M. AtCSLD3, A Cellulose Synthase-Like Gene Important for Root Hair Growth in Arabidopsis. Plant Physiol. 2001, 126, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomew, B. ISWI chromatin remodeling: One primary actor or a coordinated effort? Curr. Opin. Struct. Biol. 2014, 24, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Guo, Q.; Froehlich, J.E.; Hersh, H.L.; Zienkiewicz, A.; Howe, G.A.; Benning, C. Two Abscisic Acid-Responsive Plastid Lipase Genes Involved in Jasmonic Acid Biosynthesis in Arabidopsis thaliana. Plant Cell 2018, 30, 1006–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsthoefel, N.R.; Klag, K.A.; Simeles, B.P.; Reiter, R.; Brougham, L.; Vernon, D.M. The Arabidopsis Plant Intracellular Ras-group LRR (PIRL) Family and the Value of Reverse Genetic Analysis for Identifying Genes That Function in Gametophyte Development. Plants 2013, 2, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.; Wiles, A.M.; Sharp, J.S.; Naider, F.R.; Becker, J.M.; Stacey, G. An Oligopeptide Transporter Gene Family in Arabidopsis. Plant Physiol. 2002, 128, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Parchman, T.L.; Gompert, Z.; Mudge, J.; Schilkey, F.D.; Benkman, C.W.; Buerkle, C.A. Genome-wide association genetics of an adaptive trait in lodgepole pine: Association mapping of serotiny. Mol. Ecol. 2012, 21, 2991–3005. [Google Scholar] [CrossRef]
- Mora-Márquez, F.; García-Olivares, V.; Emerson, B.C.; López de Heredia, U. DDRADSEQTOOLS: A software package for in silico simulation and testing of double-digest RADseq experiments. Mol. Ecol. Resour. 2017, 17, 230–246. [Google Scholar] [CrossRef] [Green Version]
- Hijmans, R.; van Etten, J.; Sumner, M.; Cheng, J.; Baston, D.; Bevan, A.; Bivand, R.; Busetto, L.; Canty, M.; Fasoli, B.; et al. Raster: Geographic Data Analysis and Modeling. R Package, Version 3.6-11. 2022. Available online: https://CRAN.R-project.org/package=raster (accessed on 17 December 2022).
- Bivand, R.; Pebesma, E.J.; Gómez-Rubio, V. Applied Spatial Data Analysis with R, 2nd ed.; Springer: New York, NY, USA, 2013; ISBN 978-1-4614-7617-7. [Google Scholar] [CrossRef]
- Fick, S.E.; Hijmans, R.J. WorldClim 2: New 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Doran, A.G.; Creevey, C.J. Snpdat: Easy and Rapid Annotation of Results from De Novo SNP Discovery Projects for Model and Non-Model Organisms. BMC Bioinform. 2013, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package; R Package Version 2.6-2. 2022. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 15 May 2022).
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R Package for Genetic Analysis of Populations with Clonal, Partially Clonal, and/or Sexual Reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Knaus, B.J.; Grünwald, N.J. VCFR: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef]
- Dray, S.; Dufour, A.-B. The ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Soft. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Climate Variable | PC1 | PC2 |
|---|---|---|
| Temp | −0.467 | −0.363 |
| Isothermality | −0.378 | 0.481 |
| TempSeas | 0.445 | −0.358 |
| MTofWQ | −0.245 | −0.713 |
| MTofCQ | −0.513 | −0.009 |
| Prec | 0.343 | −0.020 |
| Environmental Factor | RDA1 | RDA2 | RDA3 |
|---|---|---|---|
| Alt | −0.015 | 0.259 | 0.155 |
| Temp | 0.051 | −0.284 | 0.031 |
| Isothermality | 0.039 | 0.032 | 0.099 |
| TempSeas | −0.102 | 0.056 | −0.079 |
| MTofWQ | −0.014 | −0.310 | −0.030 |
| MTofCQ | 0.087 | −0.193 | 0.061 |
| Prec | −0.003 | 0.170 | −0.113 |
| Region | Transect | Population Sample | N | PrA | AR | Ho | He | FIS |
|---|---|---|---|---|---|---|---|---|
| Western Sayan Mountain | A | A_h_500 | 10 | 5 | 1.257 ± 0.002 | 0.043 ± 0.001 | 0.061 ± 0.001 | 0.199 ± 0.004 ** |
| A_h_1000 | 10 | 0 | 1.275 ± 0.002 | 0.055 ± 0.001 | 0.062 ± 0.001 | 0.071 ± 0.003 ** | ||
| A_h_1500 | 10 | 0 | 1.299 ± 0.002 | 0.060 ± 0.001 | 0.068 ± 0.001 | 0.081 ± 0.003 ** | ||
| A_h_2000 | 8 | 0 | 1.250 ± 0.002 | 0.041 ± 0.001 | 0.060 ± 0.001 | 0.213 ± 0.005 ** | ||
| C | C_h_500 | 9 | 16 | 1.278 ± 0.002 | 0.054 ± 0.001 | 0.067 ± 0.001 | 0.123 ± 0.004 ** | |
| C_h_1000 | 10 | 16 | 1.269 ± 0.002 | 0.044 ± 0.001 | 0.063 ± 0.001 | 0.207 ± 0.004 ** | ||
| C_h_1500 | 10 | 0 | 1.247 ± 0.002 | 0.046 ± 0.001 | 0.057 ± 0.001 | 0.132 ± 0.004 ** | ||
| Altai Mountains | D | D_h_500 | 10 | 0 | 1.282 ± 0.002 | 0.067 ± 0.001 | 0.066 ± 0.001 | −0.007 ± 0.002 |
| D_h_1000 | 8 | 0 | 1.284 ± 0.003 | 0.066 ± 0.001 | 0.065 ± 0.001 | −0.009 ± 0.002 | ||
| D_h_1500 | 10 | 6 | 1.261 ± 0.002 | 0.061 ± 0.001 | 0.061 ± 0.001 | 0.000 ± 0.002 | ||
| D_h_2000 | 10 | 0 | 1.284 ± 0.002 | 0.066 ± 0.001 | 0.065 ± 0.001 | −0.007 ± 0.002 | ||
| E | E_h_1000 | 10 | 5 | 1.276 ± 0.002 | 0.054 ± 0.001 | 0.064 ± 0.001 | 0.108 ± 0.003 ** | |
| E_h_1500 | 9 | 0 | 1.280 ± 0.002 | 0.060 ± 0.001 | 0.066 ± 0.001 | 0.051 ± 0.003 ** | ||
| E_h_2000 | 9 | 8 | 1.271 ± 0.003 | 0.055 ± 0.001 | 0.065 ± 0.001 | 0.097 ± 0.004 ** | ||
| F | F_h_500 | 9 | 0 | 1.285 ± 0.002 | 0.067 ± 0.001 | 0.064 ± 0.001 | −0.026 ± 0.002 ** | |
| F_h_1000 | 10 | 6 | 1.270 ± 0.002 | 0.064 ± 0.001 | 0.062 ± 0.001 | −0.015 ± 0.002 * | ||
| F_h_1500 | 10 | 0 | 1.255 ± 0.002 | 0.058 ± 0.001 | 0.057 ± 0.001 | −0.005 ± 0.002 | ||
| F_h_2000 | 10 | 0 | 1.260 ± 0.002 | 0.062 ± 0.001 | 0.059 ± 0.001 | −0.026 ± 0.002 ** | ||
| Kuznetsk Alatau | G | G_h_500 | 10 | 15 | 1.305 ± 0.002 | 0.065 ± 0.001 | 0.073 ± 0.001 | 0.068 ± 0.003 ** |
| G_h_1000 | 9 | 5 | 1.295 ± 0.002 | 0.061 ± 0.001 | 0.071 ± 0.001 | 0.091 ± 0.003 ** | ||
| G_h_1500 | 10 | 33 | 1.302 ± 0.002 | 0.065 ± 0.001 | 0.074 ± 0.001 | 0.073 ± 0.003 ** | ||
| East Tuva Highlands | K | K_h_1000 | 10 | 15 | 1.283 ± 0.002 | 0.053 ± 0.001 | 0.067 ± 0.001 | 0.129 ± 0.003 ** |
| K_h_1500 | 10 | 9 | 1.300 ± 0.002 | 0.064 ± 0.001 | 0.071 ± 0.001 | 0.062 ± 0.003 ** | ||
| K_h_2000 | 10 | 4 | 1.312 ± 0.002 | 0.065 ± 0.001 | 0.072 ± 0.001 | 0.065 ± 0.004 ** | ||
| Mean | 9.6 | 5.958 ± 1.652 | 1.278 ± 0.004 | 0.058 ± 0.002 | 0.065 ± 0.001 | 0.070 ± 0.015 | ||
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation, % | F-Index |
|---|---|---|---|---|
| 761 neutral SNPs | ||||
| Among transects | 391.778 | 0.491 | 1.885 | FCT = 0.019 |
| Among populations within transects | 571.122 | 0.453 | 1.739 | FSC = 0.018 |
| Within populations | 10,693.785 | 25.097 | 96.376 | FST = 0.036 |
| Total | 11,656.685 | 26.041 | ||
| All 25,143 SNPs | ||||
| Among transects | 11,683.915 | 14.009 | 1.675 | FCT = 0.017 |
| Among populations within transects | 17,631.271 | 11.997 | 1.433 | FSC = 0.015 |
| Within populations | 347,430.578 | 810.654 | 96.892 | FST = 0.017 |
| Total | 376,745.764 | 836.661 | ||
| 550 adaptive SNPs | ||||
| Among transects | 2819.564 | 5.896 | 14.854 | FCT = 0.149 |
| Among populations within transects | 1422.569 | 2.748 | 6.923 | FSC = 0.081 |
| Within populations | 13,413.821 | 31.048 | 78.223 | FST = 0.218 |
| Total | 17,655.955 | 39.692 | ||
| SNP Dataset | PrA | AR | Ho | He | FIS |
|---|---|---|---|---|---|
| All 25,143 SNPs | 5.958 | 1.278 ± 0.004 | 0.058 ± 0.002 | 0.065 ± 0.001 | 0.070 ± 0.015 |
| 761 neutral SNPs | 0.375 | 1.282 ± 0.006 | 0.058 ± 0.002 | 0.067 ± 0.001 | 0.087 ± 0.017 |
| 550 adaptive SNPs | 0.994 | 1.364 ± 0.024 | 0.097 ± 0.007 | 0.114 ± 0.008 | 0.113 ± 0.028 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novikova, S.V.; Sharov, V.V.; Oreshkova, N.V.; Simonov, E.P.; Krutovsky, K.V. Genetic Adaptation of Siberian Larch (Larix sibirica Ledeb.) to High Altitudes. Int. J. Mol. Sci. 2023, 24, 4530. https://doi.org/10.3390/ijms24054530
Novikova SV, Sharov VV, Oreshkova NV, Simonov EP, Krutovsky KV. Genetic Adaptation of Siberian Larch (Larix sibirica Ledeb.) to High Altitudes. International Journal of Molecular Sciences. 2023; 24(5):4530. https://doi.org/10.3390/ijms24054530
Chicago/Turabian StyleNovikova, Serafima V., Vadim V. Sharov, Natalia V. Oreshkova, Evgeniy P. Simonov, and Konstantin V. Krutovsky. 2023. "Genetic Adaptation of Siberian Larch (Larix sibirica Ledeb.) to High Altitudes" International Journal of Molecular Sciences 24, no. 5: 4530. https://doi.org/10.3390/ijms24054530
APA StyleNovikova, S. V., Sharov, V. V., Oreshkova, N. V., Simonov, E. P., & Krutovsky, K. V. (2023). Genetic Adaptation of Siberian Larch (Larix sibirica Ledeb.) to High Altitudes. International Journal of Molecular Sciences, 24(5), 4530. https://doi.org/10.3390/ijms24054530