
Molecular Imprinting of Benzylpiperazine: A Comparison of the Self-Assembly and Semi-Covalent Approaches


Abstract
:1. Introduction
2. Results
2.1. The Self-Assembly Approach
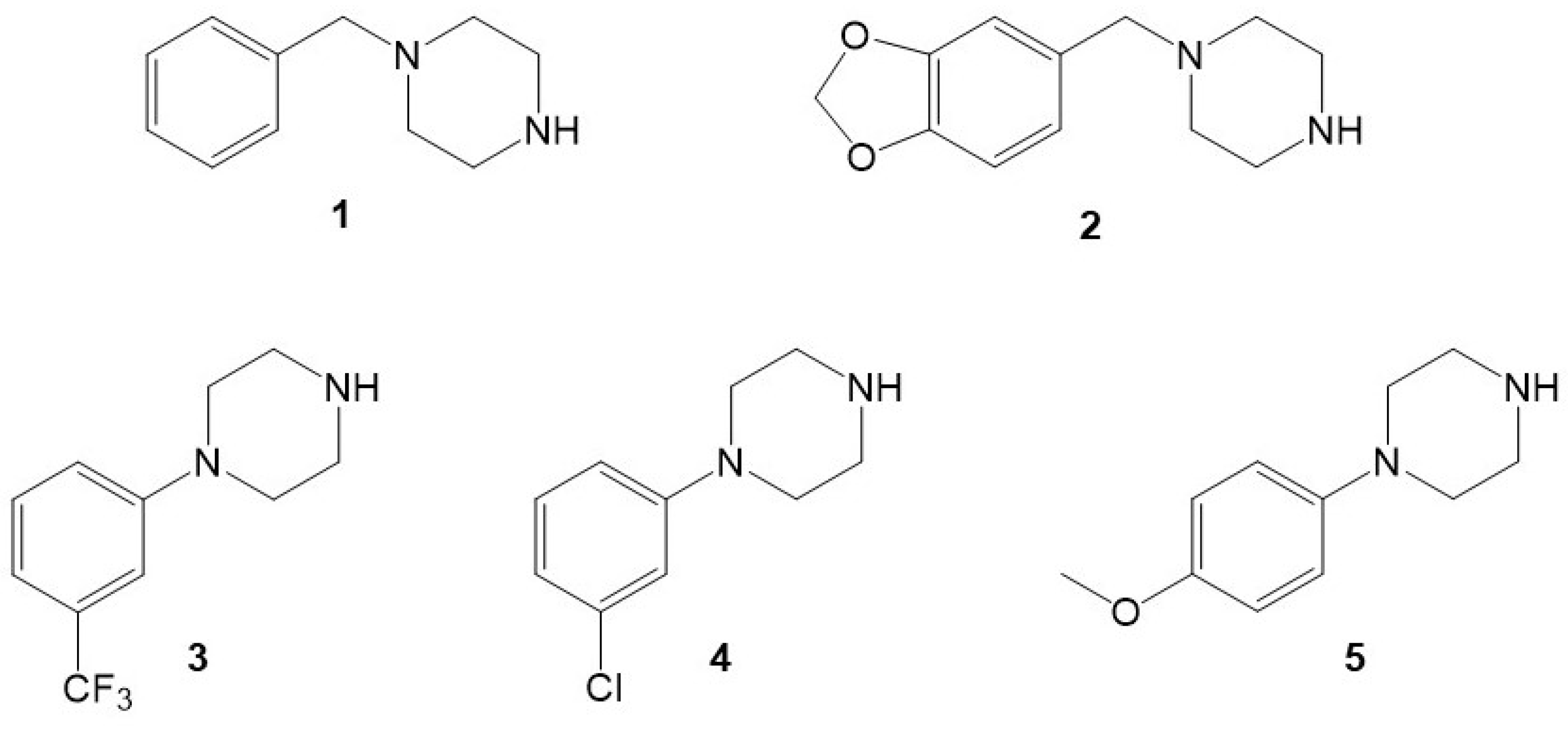
2.1.1. Template–Monomer Interaction Studies
2.1.2. Selection of Crosslinker
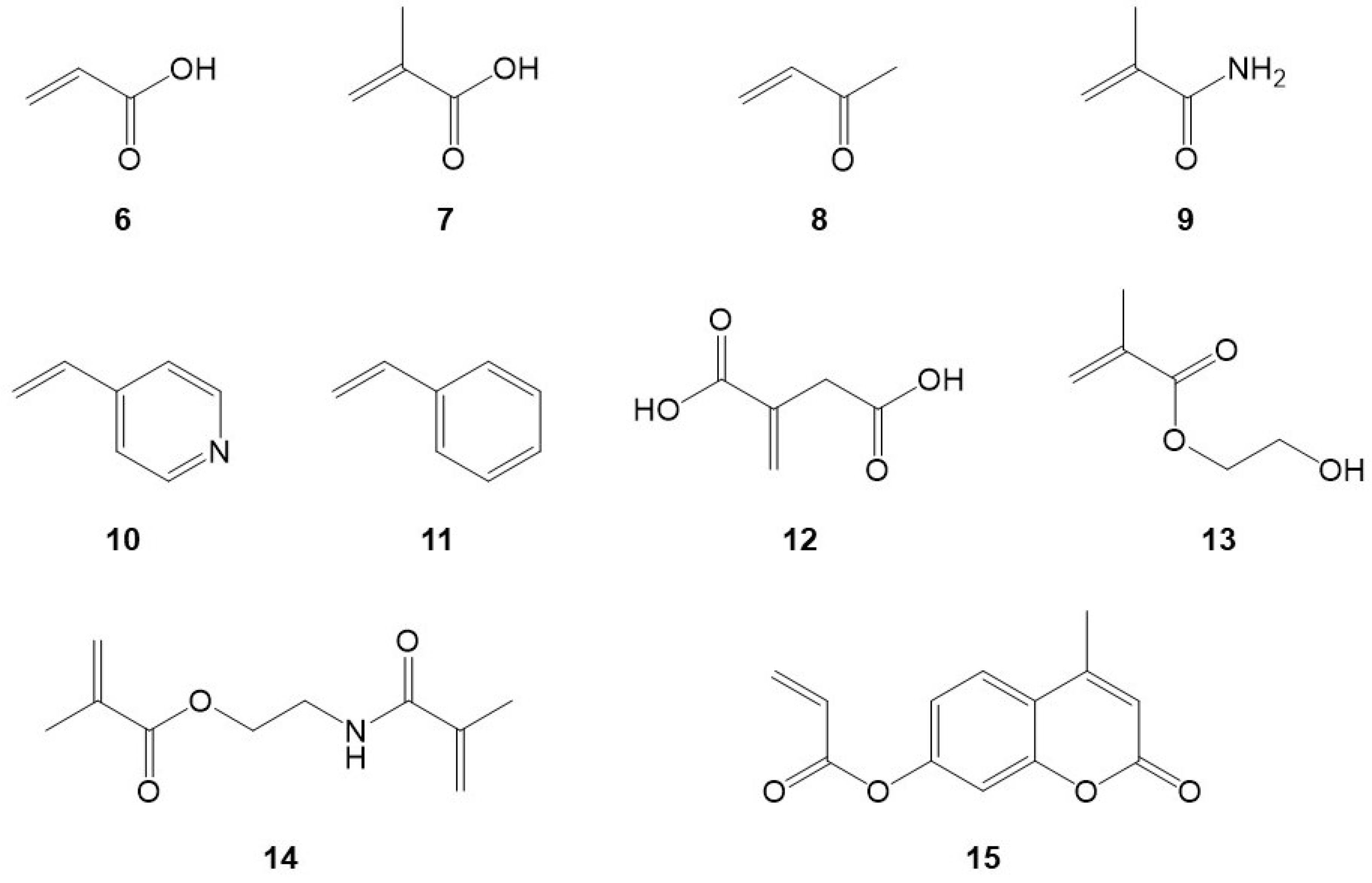
2.1.3. Preparation of 1-MIPs
2.1.4. Physical Characterisation
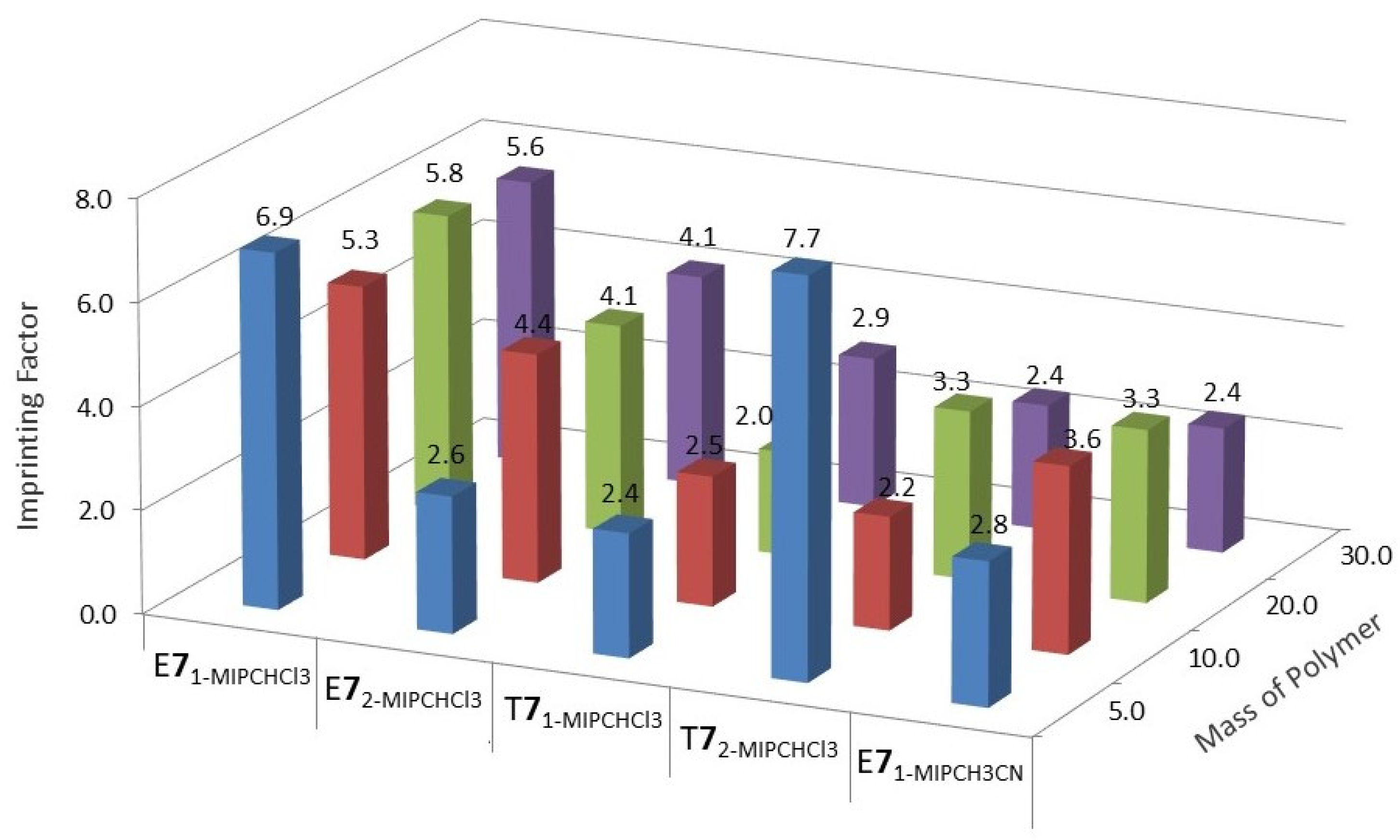
2.1.5. Evaluation of the Imprinting Effect
2.1.6. Binding Isotherms
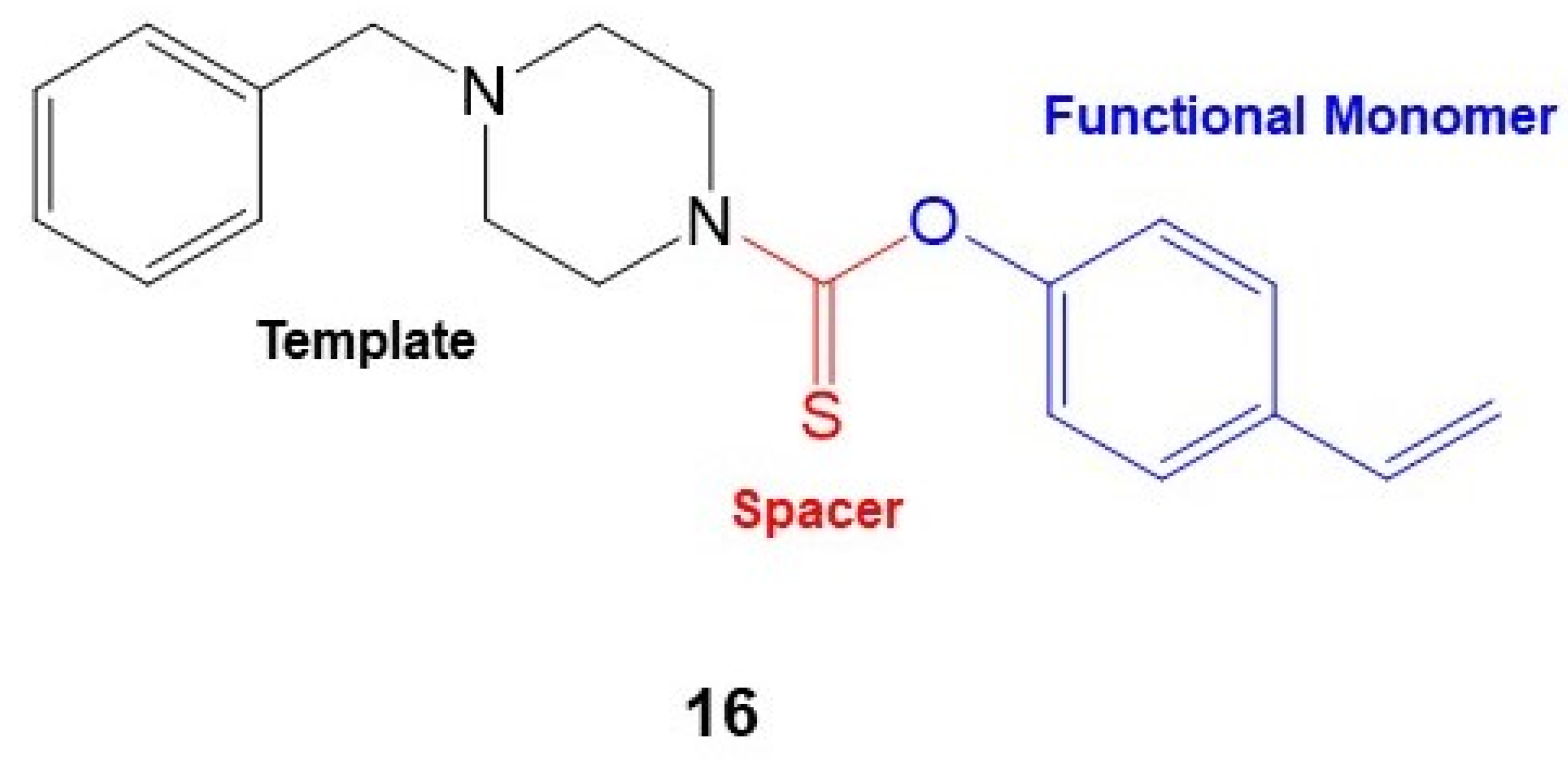
2.2. The Semi-Covalent Approach
2.2.1. Evaluation of Imprinting Effect
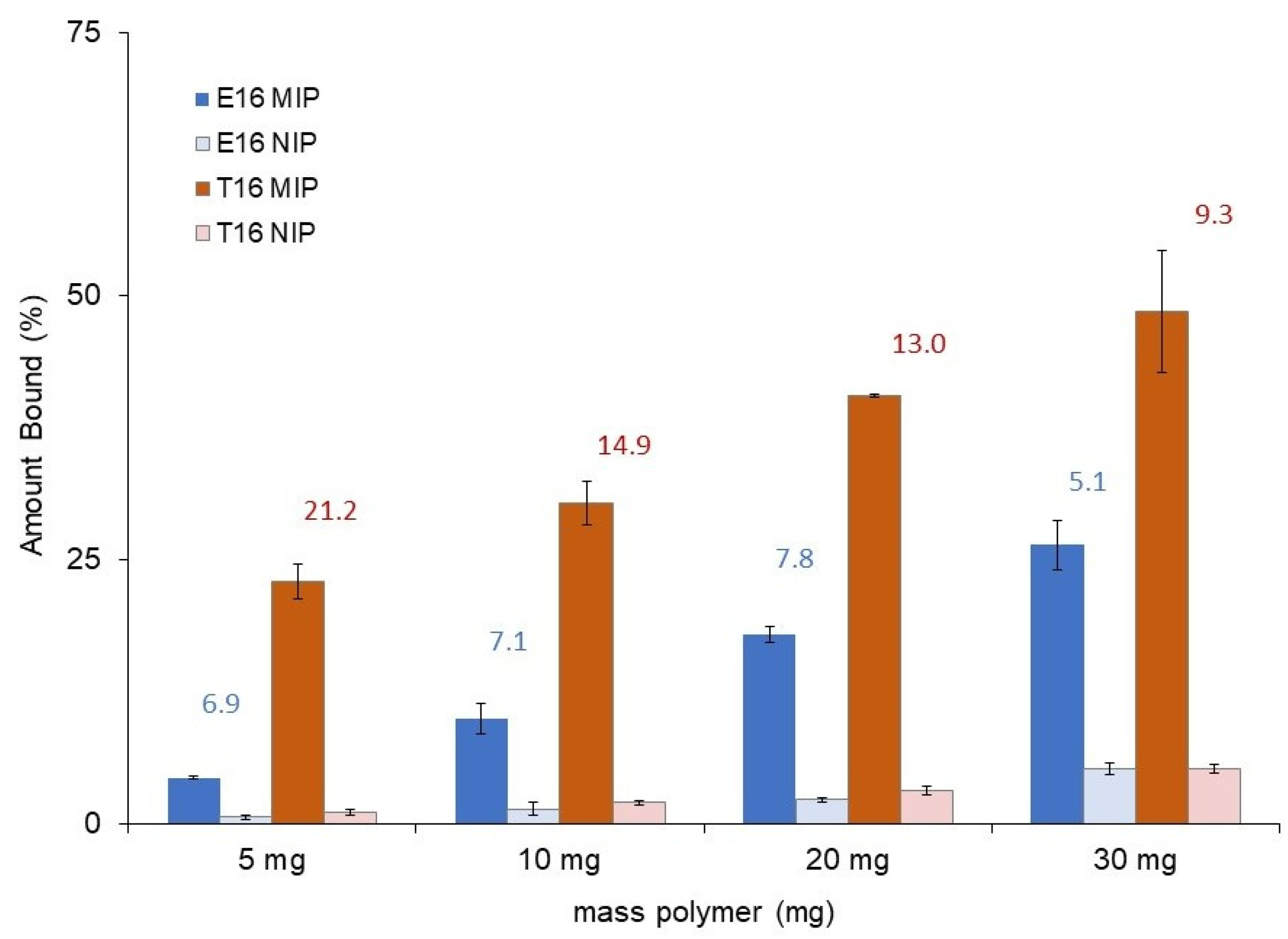
2.2.2. Binding Isotherms
2.3. Selectivity Studies
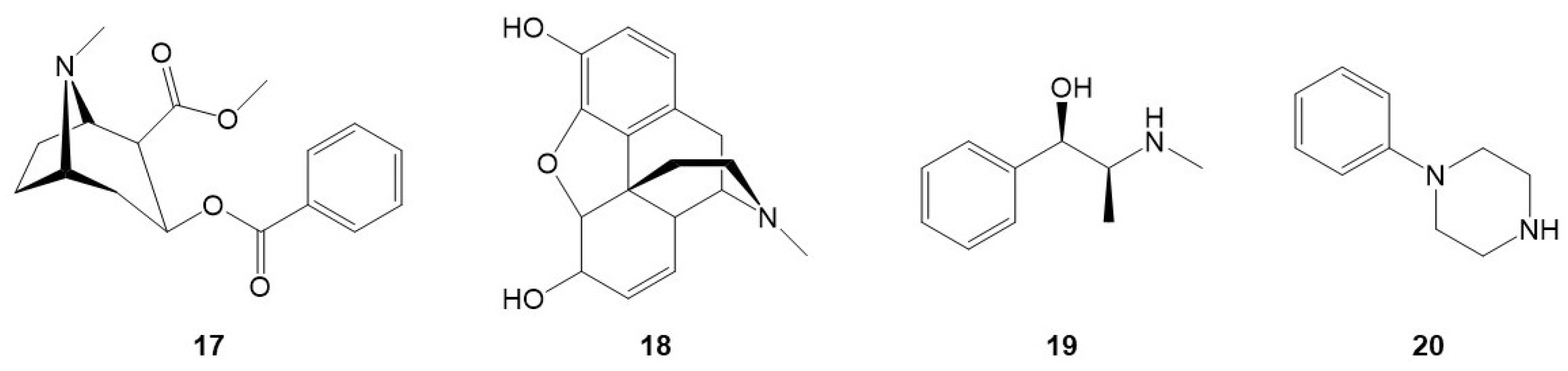
2.3.1. Cross-Reactivity Tests
2.3.2. Binary Competitive Assays
2.3.3. Selectivity of 1-MIPs: Implication on Their Applications
3. Materials and Methods
3.1. Reagents
3.2. Preparation of N,O-Bismethacryloyl Ethanolamine (NOBE, 14)
3.3. Preparation of 7-Hydroxy-4-methylcoumarin Acrylate (15)
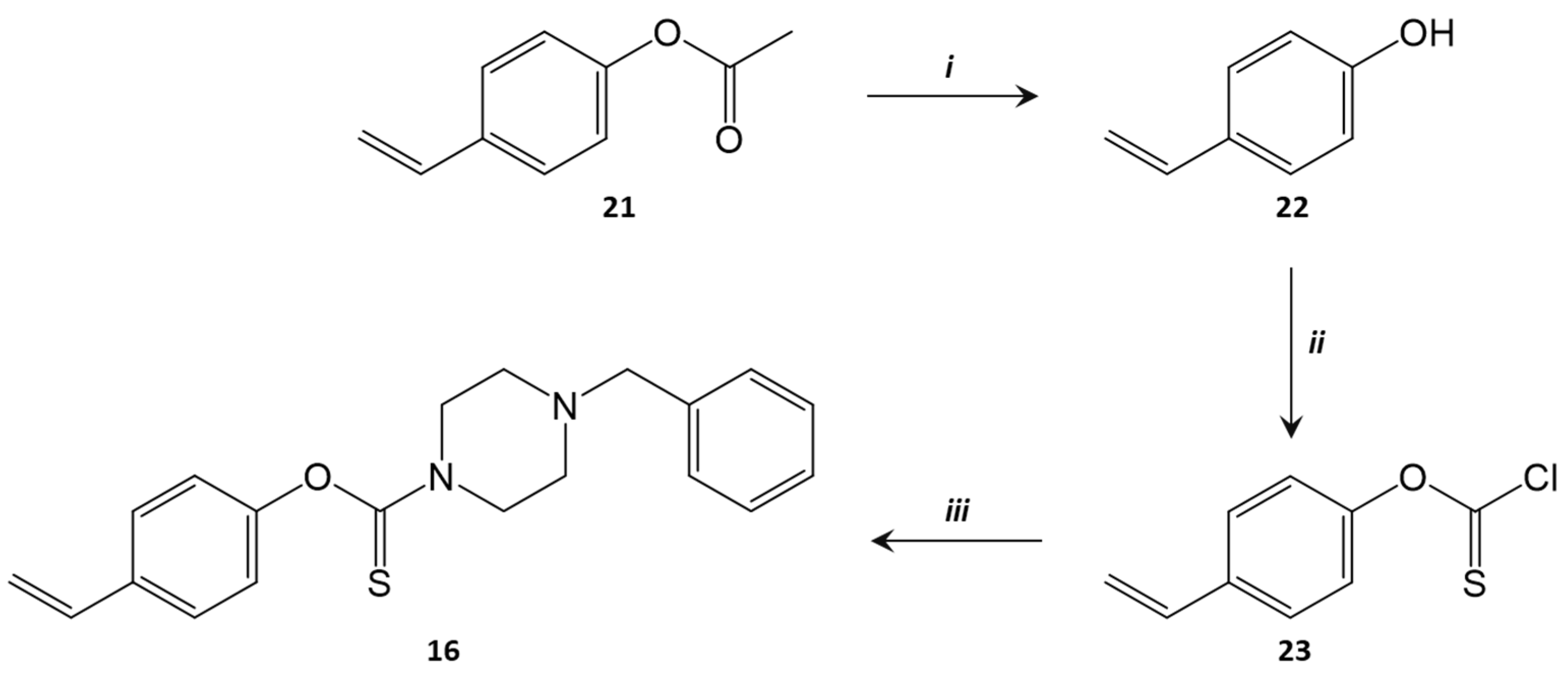
3.4. Synthesis of Benzylpiperazine (4-Vinylphenyl) Carbamate (16)
3.4.1. Synthesis of 4-Vinylphenol (22) [68]
3.4.2. Synthesis of 4-Vinylphenyl Chlorothioformate (23) [69]
3.4.3. Synthesis of O-4-Vinyl 4-Benzylpiperazine-1-carbothioate (16)
3.5. Molecular Modelling
3.6. NMR Spectroscopic Analysis
3.7. Polymer Synthesis and Template Extraction
3.7.1. Self-Assembly MIPs
3.7.2. Semi-Covalent MIPs
3.8. Batch-Binding Tests for 1
3.8.1. HPLC Analytical Method
3.8.2. Sorption Tests: Evaluation of Imprinting Effect
3.8.3. Time-Binding Study
3.8.4. Saturation Binding
3.9. Selectivity Studies
3.10. Physical Characterisation
3.10.1. Scanning Electron Microscopy
3.10.2. Swelling Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, B.M.Z.; Butler, R. BZP-party pills: A review of research on benzylpiperazine as a recreational drug. Int. J. Drug Policy 2011, 22, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bye, C.; Munro-Faure, A.D.; Peck, A.W.; Young, P.A. A comparison of the effects of 1-benzylpiperazine and dexamphetamine on human performance tests. Eur. J. Clin. Pharmacol. 1973, 6, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Campbell, H.; Cline, W.; Evans, M.; Lloyd, J.; Peck, A. Comparison of the effects of dexamphetamine and 1-benzylpiperazine in former addicts. Eur. J. Clin. Pharmacol. 1973, 6, 170–176. [Google Scholar] [CrossRef]
- Tsutsumi, H.; Katagi, M.; Miki, A.; Shima, N.; Kamata, T.; Nishikawa, M.; Nakajima, K.; Tsuchihashi, H. Development of simultaneous gas chromatography-mass spectrometric and liquid chromatography-electrospray ionization mass spectrometric determination method for the new designer drugs, N-benzylpiperazine (BZP), 1-(3-trifluoromethylphenyl)piperazine (TFMPP) and their main metabolites in urine. J. Chrom. B Anal. Tech. Biomed. Life Sci. 2005, 819, 315–322. [Google Scholar]
- Johnstone, A.C.; Lea, R.A.; Brennan, K.A.; Schenk, S.; Kennedy, M.A.; Fitzmaurice, P.S. Review: Benzylpiperazine: A drug of abuse? J. Psychopharmacol. 2007, 21, 888–894. [Google Scholar] [CrossRef]
- Gee, P.; Richardson, S.; Woltersdorf, W.; Moore, G. Toxic effects of BZP-based herbal party pills in humans: A prospective study in Christchurch, New Zealand. N. Z. Med. J. 2005, 118, U1784. [Google Scholar]
- Schechter, M.D. Serotonergic-dopaminergic mediation of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”). Pharmacol. Biochem. Behav. 1988, 31, 817–824. [Google Scholar] [CrossRef]
- Wikström, M.; Holmgren, P.; Ahlner, J. A2 (N-Benzylpiperazine) a New Drug of Abuse in Sweden. J. Anal. Toxicol. 2004, 28, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Luethi, D.; Liechti, M.E. Designer drugs: Mechanism of action and adverse effects. Arch. Toxicol. 2020, 94, 1085–1133. [Google Scholar] [CrossRef] [Green Version]
- Wilkin, C.; Sweetsur, P.; Parker, K. The impact of the prohibition of benzylpiperazine (BZP) “legal highs” on the availability, price and strength of BZP in New Zealand. Drug Alcohol Depend. 2014, 144, 47–52. [Google Scholar] [CrossRef]
- De Boer, D.; Bosman, I.J.; Hidvégi, E.; Manzoni, C.; Benkö, A.A.; dos Reys, L.J.A.L.; Maes, R.A.A. Piperazine-like compounds: A new group of designer drug-of-abuse on the European Market. Forensic Sci. Int. 2001, 121, 47–56. [Google Scholar] [CrossRef] [PubMed]
- United Nations Office on Drugs and Crime (UNODC). Recommended Methods for the Identification and Analysis of Piperazines in Seized Materials; United Nations: New York, NY, USA, 2013; pp. 1–46. Available online: https://www.unodc.org/documents/scientific/STNAR47_Piperazines_Ebook.pdf (accessed on 3 December 2022).
- Bassindale, T.A.; Berezowski, R. Quantitative analysis of hair samples for 1-benzylpiperazine (BZP) using high-performance liquid chromatography triple quadrupole mass spectrometry (LC-MS/MS) detection. Anal. Bioanal. Chem. 2011, 401, 2013–2017. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.G.; Silva, I.C.O.F.; Arantes, L.C.; Stefano, J.S.; Lima, C.D.; Melo, L.M.A.; Munoz, R.A.A.; dos Santos, W.T.P.; Richter, E.M. Simple and rapid electrochemical detection of 1-benzylpiperazine on carbon screen-printed electrode. Microchem. J. 2021, 167, 106282. [Google Scholar] [CrossRef]
- Waddell, S.A.; Fernandez, C.; Inverarity, C.C.; Prabhu, R.R. Extending the capability of forensic electrochemistry to the novel psychoactive substance benzylpiperazine. Sens. Bio-Sens. Res. 2017, 13, 28–39. [Google Scholar] [CrossRef]
- Philp, M.; Shimmon, R.; Natasha Stojanovska, N.; Tahtouhb, M.; Shanlin Fu, S. Development and validation of a presumptive colour spot test method for the detection of piperazine analogues in seized illicit materials. Anal. Methods 2013, 5, 5402–5410. [Google Scholar] [CrossRef] [Green Version]
- Holdsworth, C.I.; Bowyer, M.C.; Lennard, C.; McCluskey, A. Formulation of Cocaine-Imprinted Polymers Utilizing Molecular Modelling and NMR Analysis. Aus. J. Chem. 2005, 58, 315–320. [Google Scholar] [CrossRef]
- Brisbane, C.; McCluskey, A.; Bowyer, M.; Holdsworth, C.I. Molecularly imprinted films of acrylonitrile/methyl methacrylate/acrylic acid terpolymers: Influence of methyl methacrylate in the binding performance of L-ephedrine imprinted films. Org. Biomol. Chem. 2013, 11, 2872–2884. [Google Scholar] [CrossRef]
- Xiao, D.; Jiang, Y.; Bi, Y. Molecularly imprinted polymers for the detection of illegal drugs and additives: A review. Microchim. Acta 2018, 185, 247. [Google Scholar] [CrossRef]
- Mosbach, K.; Ramström, O. The Emerging Technique of Molecular Imprinting and Its Future Impact on Biotechnology. Nat. Biotechnol. 1996, 14, 163–170. [Google Scholar] [CrossRef]
- Herrera-Chacon, A.; Gonzalez-Calabuig, A.; del Valle, M. Dummy Molecularly Imprinted Polymers Using DNP as a Template Molecule for Explosive Sensing and Nitroaromatic Compound Discrimination. Chemosensors 2021, 9, 255. [Google Scholar] [CrossRef]
- Haupt, K.; Mosbach, K. Molecularly Imprinted Polymers and Their Use in Biomimetic Sensors. Chem. Rev. 2000, 100, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Chacón, A.; Cetó, X.; del Valle, M. Molecularly imprinted polymers—Towards electrochemical sensors and electronic tongues. Anal. Bioanal. Chem. 2021, 413, 6117–6140. [Google Scholar] [CrossRef] [PubMed]
- Scheller, F.W.; Zhang, X.; Yarman, A.; Wollenberger, U.; Gyurcsányi, R.E. Molecularly imprinted polymer-based electrochemical sensors for biopolymers. Curr. Opin. Electrochem. 2019, 14, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Liu, P.; Liu, X.; Liu, S.; Zhang, Z. Molecularly imprinted polymers for electrochemical detection and analysis: Progress and perspectives. J. Mater. Res. Technol. 2020, 9, 12568–12584. [Google Scholar] [CrossRef]
- Wulff, G.; Sarhan, A. The Use of Polymers with Enzyme-Analogous Structures for the Resolution of Racemates. Angew. Chem. Int. Ed. 1972, 11, 341–344. [Google Scholar]
- Brüggemann, O.; Haupt, K.; Ye, L.; Yilmaz, E.; Mosbach, K. New configurations and applications of molecularly imprinted polymers. J. Chromatogr. A 2000, 889, 15–24. [Google Scholar] [CrossRef]
- Bertolotti, S.G.; Previtali, C.M.; Rufs, A.M.; Encinas, M.V. Riboflavin/Triethanolamine as Photoinitiator System of Vinyl Polymerization. A Mechanistic Study by Laser Flash Photolysis. Macromolecules 1999, 32, 2920–2924. [Google Scholar] [CrossRef]
- Piletska, E.; Romero-Guerra, M.; Chianella, I.; Karim, K.; Turner, A.; Piletsky, S. Towards the development of multisensor for drugs of abuse based on molecular imprinted polymers. Anal. Chim. Acta 2005, 542, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Piletsky, S.; Karim, K.; Piletska, E.; Turner, A.; Day, C.; Freebairn, K.; Legge, C. Recognition of ephedrine enantiomers by molecularly imprinted polymers designed using a computational approach. Analyst 2001, 126, 1826–1830. [Google Scholar] [CrossRef]
- McCluskey, A.; Holdsworth, C.I.; Bowyer, M.C. Molecularly imprinted polymers (MIPs): Sensing, an explosive new opportunity? Org. Biomol. Chem. 2007, 5, 3233–3244. [Google Scholar] [CrossRef]
- Haupt, K.; Linares, A.V.; Bompart, M.; Bui, B.T. Molecularly imprinted polymers. Top. Curr. Chem. 2012, 325, 1–28. [Google Scholar] [CrossRef]
- Puoci, F.; Cirillo, G.; Curcio, M.; Parisi, O.I.; Iemma, F.; Picci, N. Molecularly imprinted polymers in drug delivery: State of art and future perspectives. Expert Opin. Drug Deliv. 2011, 8, 1379–1393. [Google Scholar] [CrossRef] [PubMed]
- Puoci, F.; Iemma, F.; Picci, N. Stimuli-responsive molecularly imprinted polymers for drug delivery: A review. Curr. Drug Deliv. 2008, 5, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Soppera, O.; Haupt, K. Photopolymerization and photostructuring of molecularly imprinted polymers for sensor applications—A review. Anal. Chim. Acta 2012, 2, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Karim, K.; Breton, F.; Rouillon, R.; Piletska, E.V.; Guerreiro, A.; Chianella, I.; Piletsky, S.A. How to find effective functional monomers for effective molecularly imprinted polymers? Adv. Drug Deliv. Rev. 2005, 57, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, S.; Piletsky, S.; Piletska, E.; Chen, B.; Karim, K.; Turner, A.P.F. Bite-and-Switch’ approach using computationally designed molecularly imprinted polymers for sensing of creatinine. Biosens. Bioelectron. 2001, 16, 631–637. [Google Scholar] [CrossRef]
- Breton, F.; Rouillon, R.; Piletska, E.V.; Karim, K.; Guerreiro, A.; Chianella, I.; Piletsky, S.A. Virtual imprinting as a tool to design efficient MIPs for photosynthesis-inhibiting herbicides. Biosens. Bioelectron. 2007, 22, 1948–1954. [Google Scholar] [CrossRef]
- Pavel., D.; Lagowski, J.; Lepage, C.J. Computationally designed monomers for molecular imprinting of chemical warfare agents—Part V. Polymer 2006, 47, 8389–8399. [Google Scholar] [CrossRef]
- Williams, D.H.; Cox, J.P.L.; Doig, A.J.; Gardner, M.; Gerhard, U.; Kaye, P.T.; Lal, A.R.; Nicholls, I.A.; Salter, C.J.; Mitchell, R.C. Toward the semiquantitative estimation of binding constants. Guides for peptide-peptide binding in aqueous solution. J. Am. Chem. Soc. 1991, 113, 7020–7030. [Google Scholar] [CrossRef]
- Nicholls, I.A.; Adbo, K.; Andersson, H.; Andersson, P.; Ankarloo, J.; Hedin-Dahlström, J.; Jokela, P.; Karlsson, J.; Olofsson, L.; Rosengren, J. Can we rationally design molecularly imprinted polymers? Anal. Chim. Acta 2001, 435, 9–18. [Google Scholar] [CrossRef]
- Sellergren, B.; Lepistoe, M.; Mosbach, K. Highly enantioselective and substrate-selective polymers obtained by molecular imprinting utilizing noncovalent interactions. NMR and chromatographic studies on the nature of recognition. J. Am. Chem. Soc. 1988, 110, 5853–5860. [Google Scholar] [CrossRef]
- Whitcombe, M.J.; Martin, L.; Vulfson, E.N. Predicting the selectivity of imprinted polymers. Chromatographia 1998, 47, 457–464. [Google Scholar] [CrossRef]
- Andersson, H.S.; Nicholls, I.A. Spectroscopic evaluation of molecular imprinting polymerization systems. Bioorg. Chem. 1997, 25, 203–211. [Google Scholar] [CrossRef]
- Ye, L.L.; Mosbach, K. Molecular Imprinting: Synthetic Materials as Substitutes for Biological Antibodies and Receptors. Chem. Mater. 2008, 20, 859–868. [Google Scholar] [CrossRef]
- Wulff, G. Biorecognition in Molecularly Imprinted Polymers. In Molecular Interactions in Bioseparations; Ngo, T.T., Ed.; Springer: Boston, MA, USA, 1993; pp. 363–381. [Google Scholar]
- Mosbach, K.; Haupt, K. Some new developments and challenges in non-covalent molecular imprinting technology. J. Mol. Recognit. 1998, 11, 62–68. [Google Scholar] [CrossRef]
- Harris, T.K.; Mildvan, A.S. High-Precision Measurement of Hydrogen Bond Lengths in Proteins by Nuclear Magnetic Resonance Methods. Proteins 1999, 35, 275–282. [Google Scholar] [CrossRef]
- Sibrian-Vazquez, M.; Spivak, D.A. Enhanced Enantioselectivity of Molecularly Imprinted Polymers Formulated with Novel Cross-Linking Monomers. Macromolecules 2003, 36, 5105–5113. [Google Scholar] [CrossRef]
- Spivak, D. Optimization, evaluation, and characterization of molecularly imprinted polymers. Adv. Drug Deliv Rev. 2005, 57, 1779–1794. [Google Scholar] [CrossRef]
- Schwarz, L.; Holdsworth, C.I.; McCluskey, A.; Bowyer, M.C. Synthesis and evaluation of a molecularly imprinted polymer selective to 2,4,6-trichlorophenol. Aust. J. Chem. 2004, 57, 759–764. [Google Scholar] [CrossRef]
- Whitcombe, M.J.; Rodriguez, M.E.; Villar, P.; Vulfson, E.N. A new method for the introduction of recognition site functionality into polymers prepared by molecular imprinting—Synthesis and characterization of polymeric receptors for cholesterol. J. Am. Chem. Soc. 1995, 117, 7105–7111. [Google Scholar] [CrossRef]
- Kirsch, N.; Alexander, C.; Davies, S.; Whitcombe, M.J. Sacrificial spacer and non-covalent routes toward the molecular imprinting of “poorly-functionalized” N-heterocycles. Anal. Chim. Acta 2004, 504, 63–71. [Google Scholar] [CrossRef]
- Chunta, S.; Sroysee, W.; Amatatongchai, M.; Lieberzeit, P.A. Molecularly Imprinted Polymers to Detect Profenofos and Carbofuran Selectively with QCM Sensors. Phys. Med. 2019, 7, 100016. [Google Scholar]
- Petcu, M.; Cooney, J.; Cook, C.; Lauren, D.; Schaare, P.; Holland, P. Molecular imprinting of a small substituted phenol of biological importance. Anal. Chim. Acta 2001, 435, 49–55. [Google Scholar] [CrossRef]
- Booker, K.; Holdsworth, C.I.; Doherty, C.M.; Hill, A.J.; Bowyer, M.C.; McCluskey, A. Ionic liquids as porogens for molecularly imprinted polymers: Propranolol, a model study. Org. Biomol. Chem. 2014, 12, 7201–7210. [Google Scholar] [CrossRef]
- Booker, K.; Bowyer, M.C.; Holdsworth, C.I.; McCluskey, A. Efficient preparation and improved sensitivity of molecularly imprinted polymers using room temperature ionic liquids. Chem. Commun. 2006, 1730–1732. [Google Scholar] [CrossRef]
- Schwarz, L.; Bowyer, M.C.; Holdsworth, C.I.; McCluskey, M.C. Synthesis and Evaluation of a Molecularly Imprinted Polymer Selective to 2,4,6-Trichloroanisole. Aust. J. Chem. 2006, 59, 129–134. [Google Scholar] [CrossRef]
- Pardo, A.; Mespouille, L.; Blankert, B.; Trouillas, P.; Surin, M.; Dubois, P.; Duez, P. Quercetin-imprinted chromatographic sorbents revisited: Optimization of synthesis and rebinding protocols for application to natural resources. J. Chromatograp. A 2014, 1364, 128–139. [Google Scholar] [CrossRef]
- Abu-Alsoud, G.F.; Hawboldt, K.A.; Bottaro, C.S. Comparison of Four Adsorption Isotherm Models for Characterizing Molecular Recognition of Individual Phenolic Compounds in Porous Tailor-Made Molecularly Imprinted Polymer Films. ACS Appl. Mater. Interfaces 2020, 12, 11998–12009. [Google Scholar] [CrossRef]
- Rajkumar, R.; Warsinke, A.; Möhwald, H.; Scheller, F.W.; Katterle, M. Analysis of recognition of fructose by imprinted polymers. Talanta 2008, 76, 1119–1123. [Google Scholar] [CrossRef]
- Muhammad, T.; Nur, Z.; Piletska, E.V.; Yimit, O.; Piletsky, S.A. Rational design of molecularly imprinted polymer: The choice of cross-linker. Analyst 2012, 137, 2623–2628. [Google Scholar] [CrossRef]
- Turner, N.W.; Holmes, N.; Brisbane, C.; McGeachie, A.B.; Bowyer, M.C.; McCluskey, A.; Holdsworth, C.I. Effect of template on the formation of phase-inversed molecularly imprinted polymer thin films: An assessment. Soft Matter 2009, 5, 3663–3671. [Google Scholar] [CrossRef]
- Cole, M.; Baron, M. Drugs of Abuse. In Crime Scene to Court: The Essentials of Forensic Science, 4th ed.; White, P.C., Ed.; The Royal Society of Chemistry UK: Cambridge, UK, 2016; pp. 401–443. [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction. BZP/Piperazines Drug Profile. Available online: https://www.emcdda.europa.eu/publications/drug-profiles/bzp (accessed on 15 February 2023).
- UN International Drug Control Programme. Drug Characterization/Impurity Profiling—Background and Concepts; United Nations: New York, NY, USA, 2001; pp. 1–19. Available online: https://digitallibrary.un.org/record/454230?ln=en (accessed on 15 February 2023).
- Patel, J.R.; Patel, K.H.; Patel, R.M. Reactivity ratio of novel acrylic copolymer by NMR spectroscopy. Colloid Polym. Sci. 2009, 287, 89–95. [Google Scholar] [CrossRef]
- Corson, B.B.; Heintzelman, W.J.; Schwartzman, L.H.; Tiefenthal, H.E.; Lokken, R.J.; Nickels, J.E.; Atwood, G.R.; Pavlik, F.J. Preparation of Vinylphenols and Isopropenylphenols. J. Org. Chem. 1958, 23, 544–549. [Google Scholar] [CrossRef]
- Oh, H.K.; Ha, J.S.; Sung, D.D.; Lee, I. Aminolysis of Aryl Chlorothionoformates with Anilines in Acetonitrile: Effects of Amine Nature and Solvent on the Mechanism. J. Org. Chem. 2004, 69, 8219–8223. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymers | Kd (μM) | Bmax (μmol/g) | Kd Ratio (NIP/MIP) | R2 | |
|---|---|---|---|---|---|
| E71-MIPCHCl3 | MIP | 0.27 ± 0.07 | 56.2 ± 5.6 | 5.0 | 0.966 |
| NIP | 1.36 ± 0.83 | 30.0 ± 10.4 | 0.884 | ||
| E72-MIPCHCl3 | MIP | 0.22 ± 0.04 | 111.7 ± 9.3 | 4.5 | 0.988 |
| NIP | 0.98 ± 0.50 | 66.2 ± 19.3 | 0.909 | ||
| T71-MIPCHCl3 | MIP | 0.60 ± 0.16 | 45.9 ± 6.0 | 2.5 | 0.933 |
| NIP | 1.52 ± 0.73 | 22.7 ± 6.3 | 0.934 | ||
| T72-MIPCHCl3 | MIP | 0.31 ± 0.07 | 90.1 ± 9.7 | 2.3 | 0.984 |
| NIP | 0.71 ± 0.26 | 35.7 ± 7.2 | 0.937 | ||
| E16MIPCHCl3 | MIP | 0.09 ± 0.02 | 16.8 ± 1.2 | - | 0.949 |
| T16MIPCHCl3 | MIP | 0.02 ± 0.01 | 21.5 ± 1.7 | - | 0.869 |
| MIP | 1 | 17 | 18 | 19 | 20 | |
|---|---|---|---|---|---|---|
| E71-MIPCHCl3 | Bound (%) 1 | 51.5 ± 3.4 | 6.7 ± 0.8 | 17.4 ± 3.1 | 74.5 ± 5.8 | 52.1 ± 4.9 |
| ΔB (%) 2 | 33.5 ± 2.9 | −1.3 ± 1.8 | 5.4 ± 3.8 | 34.8 ± 6.2 | 27.8 ± 5.5 | |
| XRF 3 | 1.00 | 0.13 | 0.34 | 1.44 | 1.01 | |
| E72-MIPCHCl3 | Bound (%) | 82.9 ± 2.2 | 12.2 ± 2.7 | 19.4 ± 7.4 | 82.9 ± 2.4 | 74.2 ± 3.9 |
| ΔB (%) | 45.6 ± 2.3 | 2.8 ± 2.1 | 3.3 ± 3.1 | 23.4 ± 2.5 | 35.3 ± 2.2 | |
| XRF | 1.00 | 0.15 | 0.23 | 1.00 | 0.90 | |
| T71-MIPCHCl3 | Bound (%) | 43.3 ± 3.4 | 3.5 ± 1.1 | 18.5 ± 2.5 | 54.9 ± 1.2 | 35.8 ± 4.0 |
| ΔB (%) | 31.4 ± 3.6 | 0.6 ± 2.3 | 9.0 ± 2.6 | 24.6 ± 1.2 | 14.2 ± 4.0 | |
| XRF | 1.00 | 0.08 | 0.43 | 1.27 | 0.83 | |
| T72-MIPCHCl3 | Bound (%) | 76.4 ± 3.4 | 11.2 ± 1.2 | 20.5 ± 4.2 | 76.8 ± 4.0 | 62.3 ± 1.8 |
| ΔB (%) | 52.1 ± 5.0 | 2.2 ± 1.2 | 7.0 ± 4.5 | 25.1 ± 4.1 | 35.0 ± 4.0 | |
| XRF | 1.00 | 0.15 | 0.27 | 1.01 | 0.82 | |
| E16MIPCHCl3 | Bound (%) | 26.3 ± 2.5 | 16.8 ± 0.7 | 11.0 ± 2.0 | 26.0 ± 2.2 | 18.9 ± 1.5 |
| ΔB (%) | 17.5 ± 3.2 | 8.9 ± 1.4 | 1.5± 2.3 | 12.1 ± 2.2 | 14.0 ± 2.2 | |
| XRF | 1.00 | 0.64 | 0.42 | 0.99 | 0.72 | |
| T16MIPCHCl3 | Bound (%) | 53.0 ± 1.4 | 45.0 ± 1.5 | 16.4 ± 2.2 | 39.8 ± 2.2 | 34.8 ± 4.4 |
| ΔB (%) | 48.5 ± 1.9 | 41.0 ± 1.6 | 8.5 ± 2.3 | 36.8 ± 4.6 | 32.1 ± 4.5 | |
| XRF | 1.00 | 0.85 | 0.31 | 0.75 | 0.66 |
| Polymer | Binding 1 | MIP | NIP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 17 | 18 | 19 | 1 | 17 | 18 | 19 | ||
| E71-MIPCHCl3 | Reference (%) 2 | 51.5 ± 3.4 | 18.0 ± 3.6 | ||||||
| Normalised 3 | 1.09 | 0.19 | 1.19 | 0.41 | |||||
| 1.05 | 0.38 | 0.77 | 0.38 | ||||||
| 0.94 | 1.73 | 0.66 | 2.50 | ||||||
| SI 4 | 1.00 | 0.18 | 0.37 | 1.85 | 1.00 | 0.34 | 0.49 | 3.80 | |
| E72-MIPCHCl3 | Reference (%) | 82.9 ± 2.2 | 37.2 ± 3.1 | ||||||
| Normalised | 0.93 | 0.15 | 1.11 | 0.16 | |||||
| 0.86 | 0.22 | 0.88 | 0.02 | ||||||
| 0.86 | 1.06 | 0.90 | 1.89 | ||||||
| SI | 1.00 | 0.16 | 0.26 | 1.23 | 1.00 | 0.14 | 0.02 | 2.11 | |
| T71-MIPCHCl3 | Reference (%) | 43.3 ± 3.4 | 11.9 ± 1.2 | ||||||
| Normalised | 1.12 | 0.18 | 1.90 | 0.56 | |||||
| 1.24 | 0.20 | 1.64 | 0.56 | ||||||
| 0.74 | 1.29 | 1.15 | 3.15 | ||||||
| SI | 1.00 | 0.16 | 0.16 | 1.74 | 1.00 | 0.30 | 0.34 | 2.74 | |
| T72-MIPCHCl3 | Reference (%) | 76.4 ± 3.4 | 24.3 ± 3.6 | ||||||
| Normalised | 0.89 | 0.11 | 1.11 | 0.10 | |||||
| 0.89 | 0.20 | 1.38 | 0.26 | ||||||
| 0.78 | 1.04 | 0.92 | 2.28 | ||||||
| SI | 1.00 | 0.12 | 0.23 | 1.32 | 1.00 | 0.09 | 0.19 | 2.47 | |
| E16MIPCHCl3 | Reference (%) | 26.3 ± 2.5 | 8.7 ± 2.0 | ||||||
| Normalised | 1.01 | 0.07 | 0.72 | 0.37 | |||||
| 0.33 | 0.15 | 0.35 | 0.20 | ||||||
| 0.63 | 1.14 | 0.17 | 1.54 | ||||||
| SI | 1.00 | 0.07 | 0.46 | 1.81 | 1.00 | 0.51 | 0.59 | 8.85 | |
| T16MIPCHCl3 | Reference (%) | 53.0 ± 1.4 | 4.5 ± 1.3 | ||||||
| Normalised | 0.71 | 0.13 | 0.89 | 0.60 | |||||
| 0.33 | 0.20 | 0.63 | 0.66 | ||||||
| 0.42 | 0.57 | 0.34 | 1.18 | ||||||
| SI | 1.00 | 0.18 | 0.61 | 1.34 | 1.00 | 0.68 | 1.05 | 3.50 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, K.M.; Bowyer, M.C.; McCluskey, A.; Holdsworth, C.I. Molecular Imprinting of Benzylpiperazine: A Comparison of the Self-Assembly and Semi-Covalent Approaches. Int. J. Mol. Sci. 2023, 24, 5117. https://doi.org/10.3390/ijms24065117
Wright KM, Bowyer MC, McCluskey A, Holdsworth CI. Molecular Imprinting of Benzylpiperazine: A Comparison of the Self-Assembly and Semi-Covalent Approaches. International Journal of Molecular Sciences. 2023; 24(6):5117. https://doi.org/10.3390/ijms24065117
Chicago/Turabian StyleWright, Kathleen M., Michael C. Bowyer, Adam McCluskey, and Clovia I. Holdsworth. 2023. "Molecular Imprinting of Benzylpiperazine: A Comparison of the Self-Assembly and Semi-Covalent Approaches" International Journal of Molecular Sciences 24, no. 6: 5117. https://doi.org/10.3390/ijms24065117
APA StyleWright, K. M., Bowyer, M. C., McCluskey, A., & Holdsworth, C. I. (2023). Molecular Imprinting of Benzylpiperazine: A Comparison of the Self-Assembly and Semi-Covalent Approaches. International Journal of Molecular Sciences, 24(6), 5117. https://doi.org/10.3390/ijms24065117