
Towards a Better Understanding of the Complexities of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID

Abstract
:1. Background
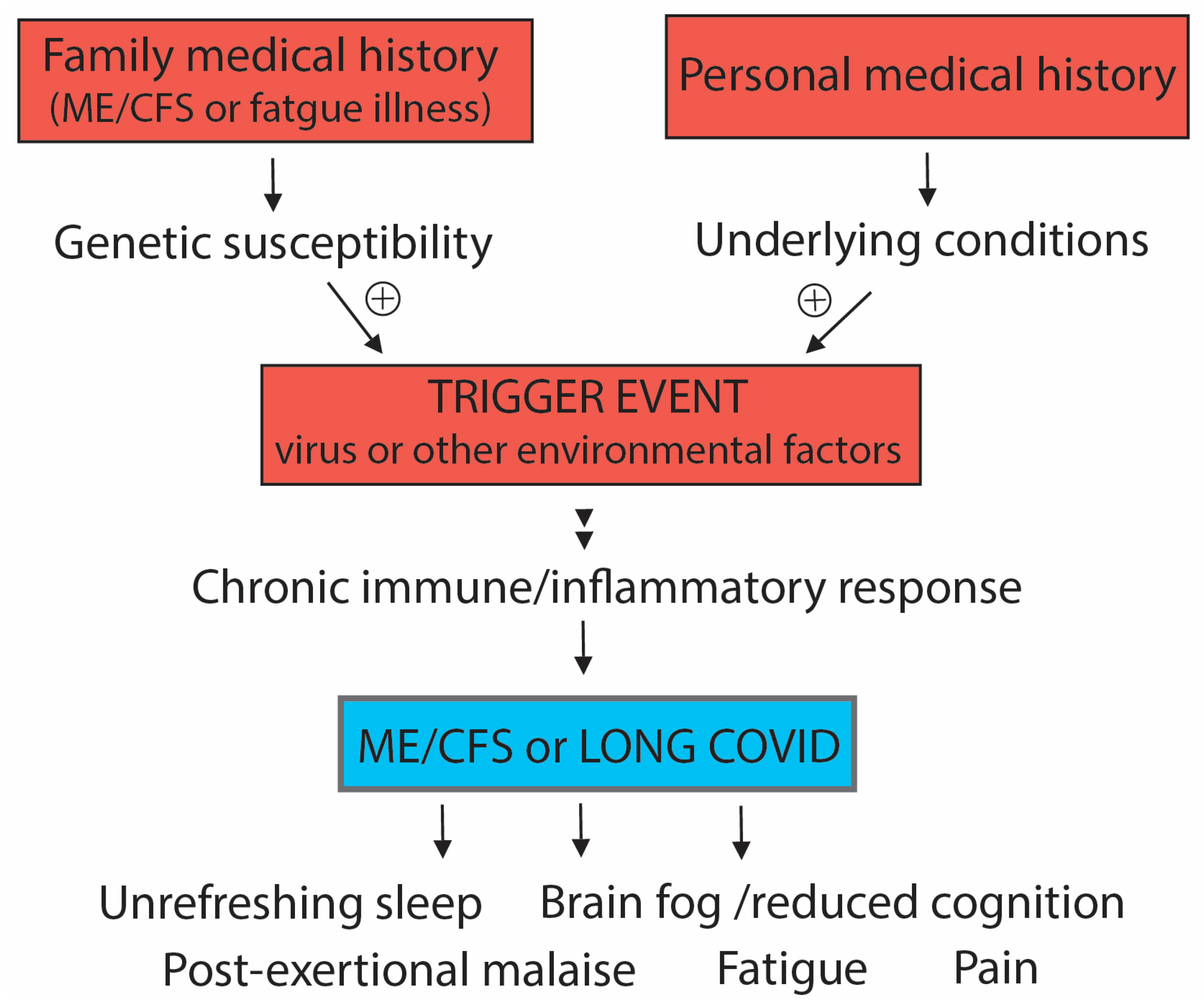
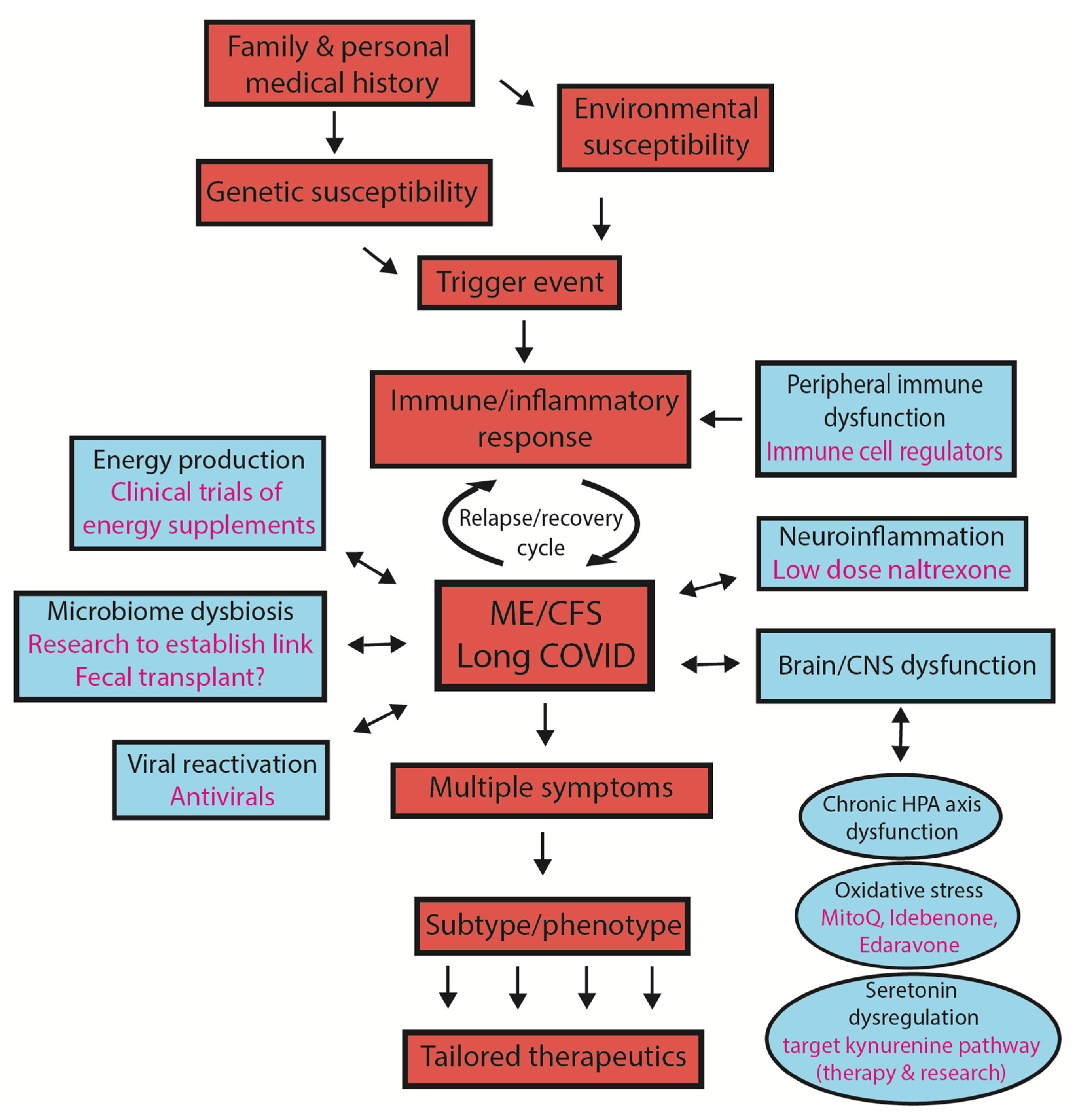
2. Susceptibility to Developing ME/CFS or Long COVID
3. What Is the Significance of ‘Subtypes/Phenotypes’ in ME/CFS?
- The concept of subtypes
- Why might subtypes be important?
- Medications
4. The Search for a Simple Universal Diagnostic Test for ME/CFS
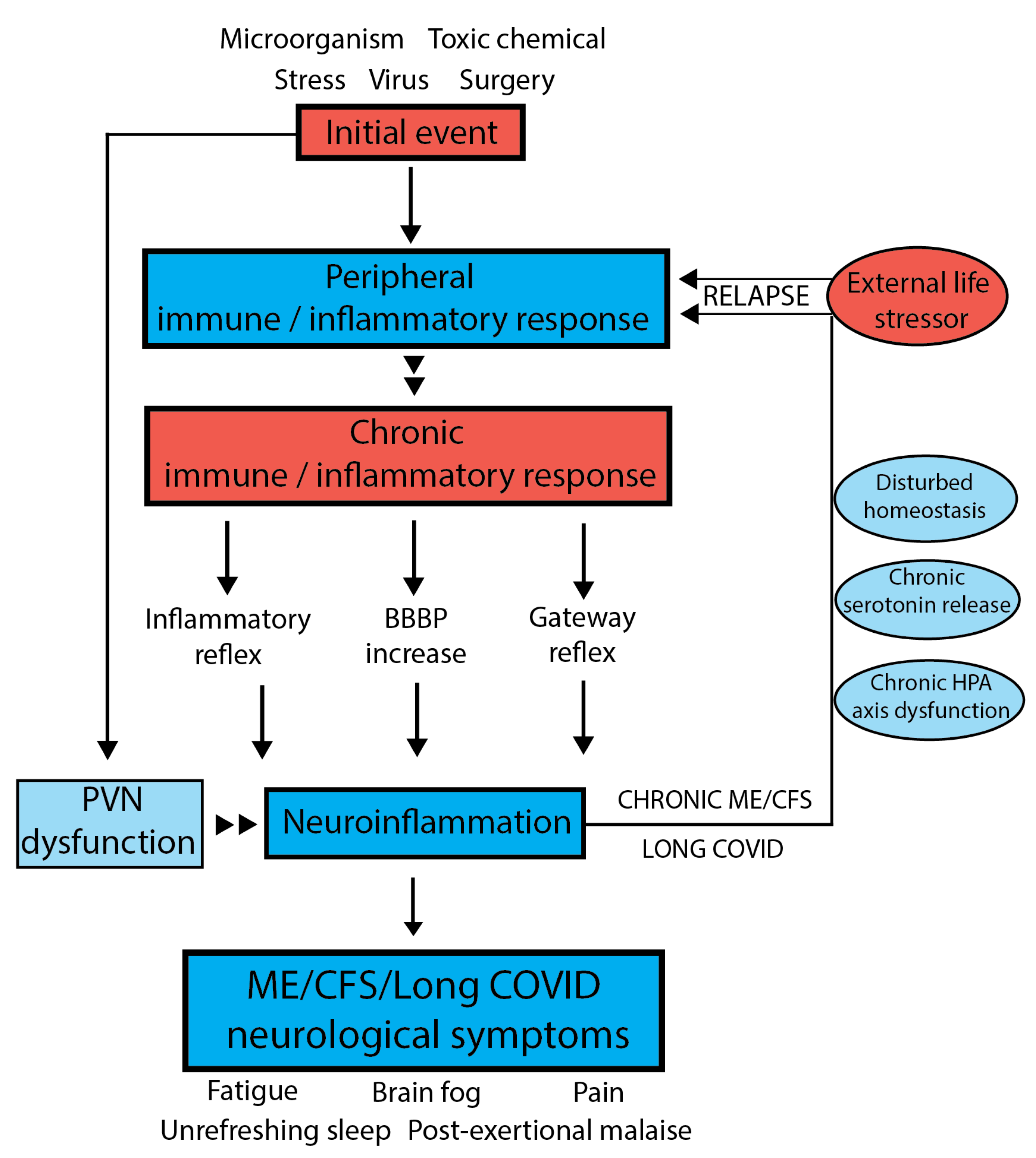
5. Holistic Model to Describe Both ME/CFS and Long COVID
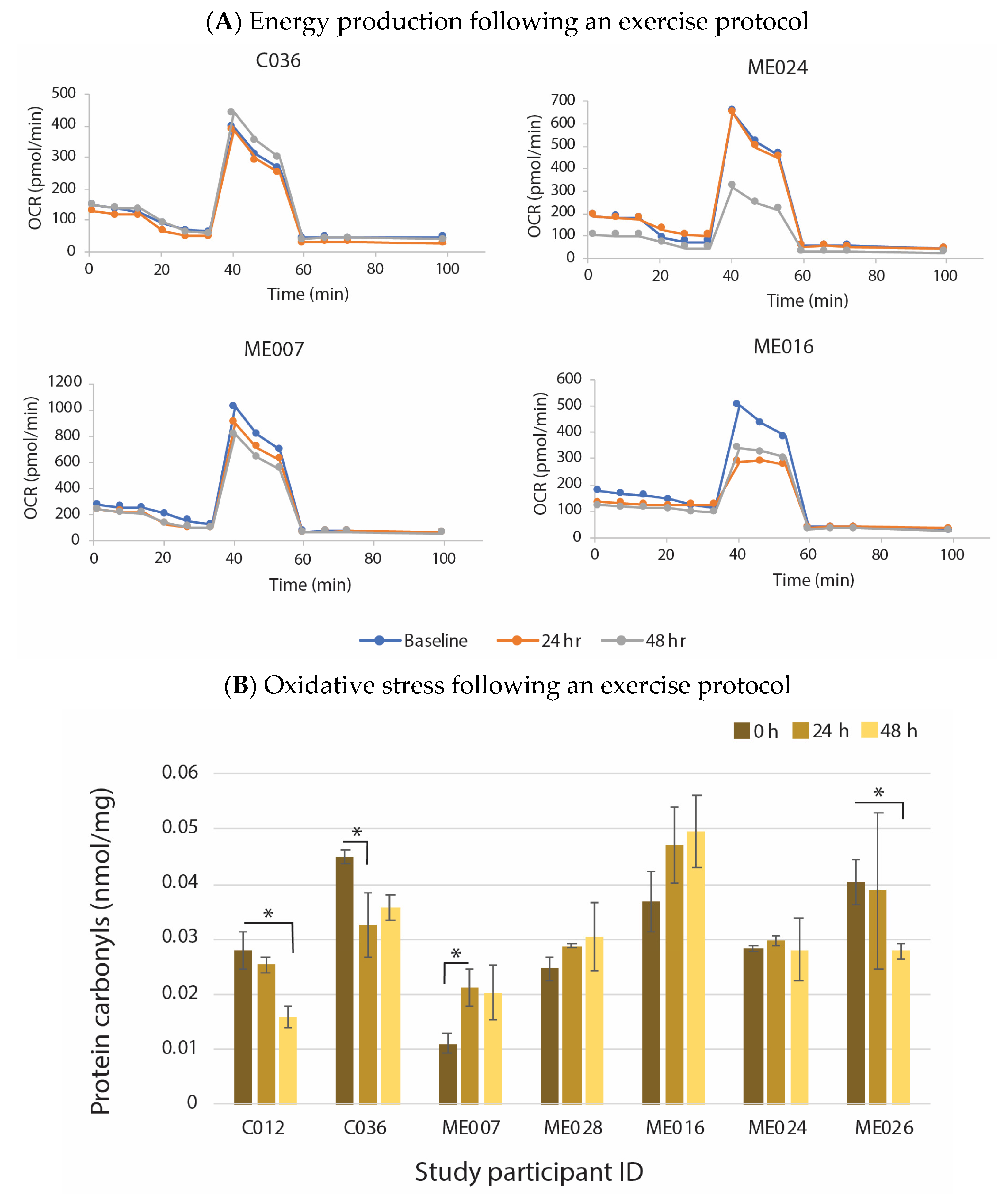
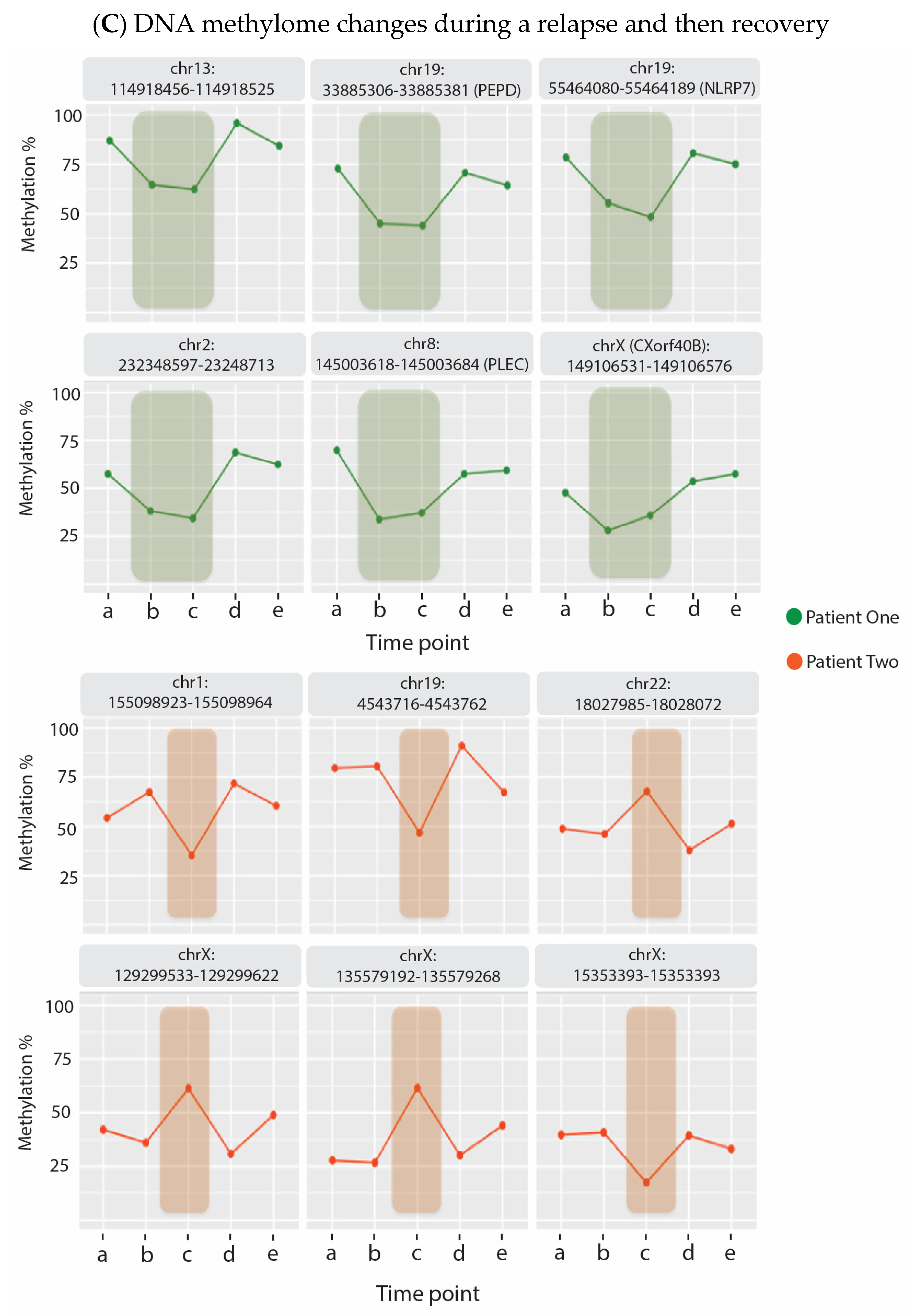
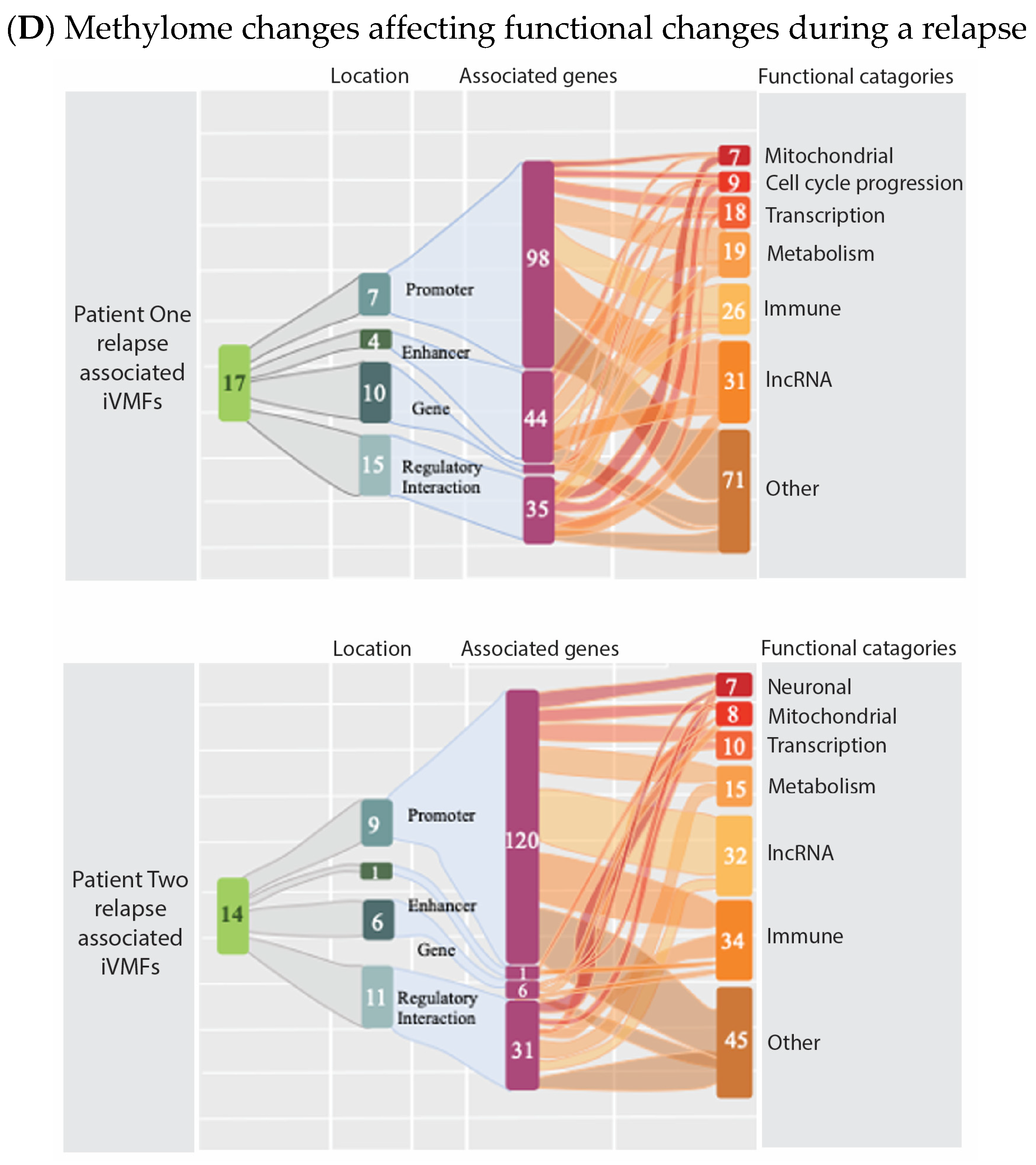
6. Evidence That Neuroinflammation Is Associated with ME/CFS
7. Underlying Mechanisms Supporting a Disturbed Homeostasis
- The inflammatory reflex:
- The Hypothalamus–Pituitary–Adrenal (HPA) axis:
- Serotonin regulation:
8. The Importance of Blood–Brain Barrier Permeability
9. Can Viral Reactivation Affect ME/CFS?
10. Can the Microbiome State Be Manipulated to Improve ME/CFS Pathology?
11. Can the Focus on Long COVID Research Provide Benefit for ME/CFS Patients?
- Longitudinal studies:
- Therapeutic possibilities:
- Clinical trials and new therapeutics
12. Patient Lifestyle Self-Management of ME/CFS and Long COVID
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, M.O.M.; Hall, K.H.; Peppercorn, K.; Tate, W.P. The significance of oxidative stress in the pathophysiology of Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Med. Res. Arch. 2022, 10, 1–22. [Google Scholar] [CrossRef]
- Lidbury, B.A.; Fisher, P.R. Biomedical Insights that Inform the Diagnosis of ME/CFS. Diagnostics 2020, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Komaroff, A.L.; Lipkin, W.I. Insights from myalgic encephalomyelitis/chronic fatigue syndrome may help unravel the pathogenesis of postacute COVID-19 syndrome. Trends Mol. Med. 2021, 27, 895–906. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- White, P.D.; Thomas, J.M.; Amess, J.; Crawford, D.H.; Grover, S.A.; Kangro, H.O.; Clare, A.W. Incidence, risk and prognosis of acute and chronic fatigue syndromes and psychiatric disorders after glandular fever. Br. J. Psychiatry 1998, 173, 475–481. [Google Scholar] [CrossRef]
- O’Mahoney, L.L.; Routen, A.; Gillies, C.; Ekezie, W.; Welford, A.; Zhang, A.; Karamchandani, U.; Simms-Williams, N.; Cassambai, S.; Ardavani, A.; et al. The prevalence and long-term health effects of Long Covid among hospitalised and non-hospitalised populations: A systematic review and meta-analysis. eClinicalMedicine 2023, 55, 101762. [Google Scholar] [CrossRef]
- Perlis, R.H.; Santillana, M.; Ognyanova, K.; Safarpour, A.; Lunz Trujillo, K.; Simonson, M.D.; Green, J.; Quintana, A.; Druckman, J.; Baum, M.A.; et al. Prevalence and Correlates of Long COVID Symptoms Among US Adults. JAMA Netw. Open 2022, 5, e2238804. [Google Scholar] [CrossRef]
- Sugiyama, A.; Miwata, K.; Kitahara, Y.; Okimoto, M.; Abe, K.; Bunthen, E.; Ouoba, S.; Akita, T.; Tanimine, N.; Ohdan, H.; et al. Long COVID occurrence in COVID-19 survivors. Sci. Rep. 2022, 12, 6039. [Google Scholar] [CrossRef]
- Antonelli, M.; Penfold, R.S.; Merino, J.; Sudre, C.H.; Molteni, E.; Berry, S.; Canas, L.S.; Graham, M.S.; Klaser, K.; Modat, M.; et al. Risk factors and disease profile of post-vaccination SARS-CoV-2 infection in UK users of the COVID Symptom Study app: A prospective, community-based, nested, case-control study. Lancet Infect. Dis. 2022, 22, 43–55. [Google Scholar] [CrossRef]
- Johnson, C. Counterpoint: Yes, Long COVID is Helping Chronic Fatigue Syndrome (ME/CFS)… Health Rising Blog. 2022. Available online: https://www.healthrising.org/blog/2022/10/25/counterpoint-long-covid-chronic-fatigue-syndrome/ (accessed on 22 January 2023).
- Friedman, K.J.; Murovska, M.; Pheby, D.F.H.; Zalewski, P. Our Evolving Understanding of ME/CFS. Medicina 2021, 57, 200. [Google Scholar] [CrossRef] [PubMed]
- Various. Topical Collection “Why Some Patients Never Fully Recover: Post Active Phase of Infection Syndromes (PAPIS)”. Healthcare 2023. Available online: https://www.mdpi.com/journal/healthcare/topical_collections/PAPIS (accessed on 22 January 2023).
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef]
- Blair, A. A Quantitative Investigation into the Personal and Family Health Histories of Long COVID and ME/CFS Patients: Identifying Susceptibility Factors and Support Needs; University of Technology Sydney: Ultimo, NSW, Australia, 2022. [Google Scholar]
- Wong, T.L.; Weitzer, D.J. Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina 2021, 57, 418. [Google Scholar] [CrossRef]
- Michelen, M.; Manoharan, L.; Elkheir, N.; Cheng, V.; Dagens, A.; Hastie, C.; O’Hara, M.; Suett, J.; Dahmash, D.; Bugaeva, P.; et al. Characterising long COVID: A living systematic review. BMJ Glob. Health 2021, 6, e005427. [Google Scholar] [CrossRef]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long COVID—Mechanisms, risk factors, and management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef]
- Hunt, J.; Blease, C.; Geraghty, K.J. Long Covid at the crossroads: Comparisons and lessons from the treatment of patients with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J. Health Psychol. 2022, 27, 3106–3120. [Google Scholar] [CrossRef]
- Sukocheva, O.A.; Maksoud, R.; Beeraka, N.M.; Madhunapantula, S.V.; Sinelnikov, M.; Nikolenko, V.N.; Neganova, M.E.; Klochkov, S.G.; Amjad Kamal, M.; Staines, D.R.; et al. Analysis of post COVID-19 condition and its overlap with myalgic encephalomyelitis/chronic fatigue syndrome. J. Adv. Res. 2022, 40, 179–196. [Google Scholar] [CrossRef]
- Gherardi, R.K.; Crépeaux, G.; Authier, F.J. Myalgia and chronic fatigue syndrome following immunization: Macrophagic myofasciitis and animal studies support linkage to aluminum adjuvant persistency and diffusion in the immune system. Autoimmun. Rev. 2019, 18, 691–705. [Google Scholar] [CrossRef]
- Cameron, B.; Flamand, L.; Juwana, H.; Middeldorp, J.; Naing, Z.; Rawlinson, W.; Ablashi, D.; Lloyd, A. Serological and virological investigation of the role of the herpesviruses EBV, CMV and HHV-6 in post-infective fatigue syndrome. J. Med. Virol. 2010, 82, 1684–1688. [Google Scholar] [CrossRef]
- Hatcher, S.; House, A. Life events, difficulties and dilemmas in the onset of chronic fatigue syndrome: A case-control study. Psychol. Med. 2003, 33, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Schlauch, K.A.; Khaiboullina, S.F.; De Meirleir, K.L.; Rawat, S.; Petereit, J.; Rizvanov, A.A.; Blatt, N.; Mijatovic, T.; Kulick, D.; Palotás, A.; et al. Genome-wide association analysis identifies genetic variations in subjects with myalgic encephalomyelitis/chronic fatigue syndrome. Transl. Psychiatry 2016, 6, e730. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.K.; Fang, H.; Whistler, T.; Unger, E.R.; Rajeevan, M.S. Convergent genomic studies identify association of GRIK2 and NPAS2 with chronic fatigue syndrome. Neuropsychobiology 2011, 64, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Dibble, J.J.; McGrath, S.J.; Ponting, C.P. Genetic risk factors of ME/CFS: A critical review. Hum. Mol. Genet. 2020, 29, R117–R124. [Google Scholar] [CrossRef]
- Hajdarevic, R.; Lande, A.; Mehlsen, J.; Rydland, A.; Sosa, D.D.; Strand, E.B.; Mella, O.; Pociot, F.; Fluge, Ø.; Lie, B.A.; et al. Genetic association study in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) identifies several potential risk loci. Brain Behav. Immun. 2022, 102, 362–369. [Google Scholar] [CrossRef]
- Das, S.; Taylor, K.; Kozubek, J.; Sardell, J.; Gardner, S. Genetic risk factors for ME/CFS identified using combinatorial analysis. J. Transl. Med. 2022, 20, 598. [Google Scholar] [CrossRef]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.P.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Chronic Fatigue Syndr. 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.P.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Mackay, A.; Tate, W.P. A compromised paraventricular nucleus within a dysfunctional hypothalamus: A novel neuroinflammatory paradigm for ME/CFS. Int. J. Immunopathol. Pharmacol. 2018, 32, 2342. [Google Scholar] [CrossRef] [Green Version]
- Murga, I.; Aranburu, L.; Gargiulo, P.A.; Gómez Esteban, J.C.; Lafuente, J.V. Clinical Heterogeneity in ME/CFS. A Way to Understand Long-COVID19 Fatigue. Front. Psychiatry 2021, 12, 735784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zang, C.; Xu, Z.; Zhang, Y.; Xu, J.; Bian, J.; Morozyuk, D.; Khullar, D.; Zhang, Y.; Nordvig, A.S.; et al. Data-driven identification of post-acute SARS-CoV-2 infection subphenotypes. Nat. Med. 2023, 29, 226–235. [Google Scholar] [CrossRef]
- Schacterle, R.S.; Komaroff, A.L. A comparison of pregnancies that occur before and after the onset of chronic fatigue syndrome. Arch. Intern. Med. 2004, 164, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruun, K.D.; Amris, K.; Vaegter, H.B.; Blichfeldt-Eckhardt, M.R.; Holsgaard-Larsen, A.; Christensen, R.; Toft, P. Low-dose naltrexone for the treatment of fibromyalgia: Protocol for a double-blind, randomized, placebo-controlled trial. Trials 2021, 22, 804. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.D.; Kalanidhi, S.; Bonilla, A.; Subramanian, A.; Ballon, J.S.; Bonilla, H. Off label use of Aripiprazole shows promise as a treatment for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A retrospective study of 101 patients treated with a low dose of Aripiprazole. J. Transl. Med. 2021, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Mitoma, F.; Turgonyi, E.; Kumar, A.; Lim, W.; Larocque, L.; Hyde, B.M. Clinical Improvement in Chronic Fatigue Syndrome Is Associated with Enhanced Natural Killer Cell-Mediated Cytotoxicity: The Results of a Pilot Study with Isoprinosine®. J. Chronic Fatigue Syndr. 2003, 11, 71–95. [Google Scholar] [CrossRef]
- Kerr, J.R.; Petty, R.; Burke, B.; Gough, J.; Fear, D.; Sinclair, L.I.; Mattey, D.L.; Richards, S.C.; Montgomery, J.; Baldwin, D.A.; et al. Gene expression subtypes in patients with chronic fatigue syndrome/myalgic encephalomyelitis. J. Infect. Dis. 2008, 197, 1171–1184. [Google Scholar] [CrossRef]
- Stoothoff, J.; Gleason, K.; McManimen, S.; Thorpe, T.; Jason, L.A. Subtyping Patients with Myalgic Encephalomyelitis (ME) and Chronic Fatigue Syndrome (CFS) By Course of Illness. J. Biosens. Biomark. Diagn. 2017, 2, 1–17. [Google Scholar] [CrossRef]
- Nacul, L.; O’Boyle, S.; Palla, L.; Nacul, F.E.; Mudie, K.; Kingdon, C.C.; Cliff, J.M.; Clark, T.G.; Dockrell, H.M.; Lacerda, E.M. How Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Progresses: The Natural History of ME/CFS. Front. Neurol. 2020, 11, 826. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, A.M.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Dynamic Epigenetic Changes during a Relapse and Recovery Cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2022, 23, 11852. [Google Scholar] [CrossRef]
- Tomas, C.; Brown, A.; Strassheim, V.; Elson, J.L.; Newton, J.; Manning, P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 2017, 12, e0186802. [Google Scholar] [CrossRef] [Green Version]
- ANZMES. ANZMES Preliminary Survey Findings. 2021. Available online: https://anzmes.org.nz/anzmes-preliminary-survey-findings/ (accessed on 22 January 2023).
- Johnson, C. Study Suggests Long COVID is Becoming More like Chronic Fatigue Syndrome (ME/CFS). Health Rising Blog. 2021. Available online: https://www.healthrising.org/blog/2021/05/17/long-covid-symptoms-chronic-fatigue-syndrome/ (accessed on 22 January 2023).
- Brurberg, K.G.; Fønhus, M.S.; Larun, L.; Flottorp, S.; Malterud, K. Case definitions for chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME): A systematic review. BMJ Open 2014, 4, e003973. [Google Scholar] [CrossRef]
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A. The chronic fatigue syndrome: A comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.S.; Bradley, A.S.; Bishop, K.N.; Kiani-Alikhan, S.; Ford, B. Chronic fatigue syndrome, the immune system and viral infection. Brain Behav. Immun. 2012, 26, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; Board on the Health of Select Populations; Institute of Medicine. The National Academies Collection: Reports funded by National Institutes of Health. In Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness; National Academies Press (US): Washington, DC, USA, 2015. [Google Scholar] [CrossRef]
- Esfandyarpour, R.; Kashi, A.; Nemat-Gorgani, M.; Wilhelmy, J.; Davis, R.W. A nanoelectronics-blood-based diagnostic biomarker for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Proc. Natl. Acad. Sci. USA 2019, 116, 10250–10257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missailidis, D.; Annesley, S.J.; Allan, C.Y.; Sanislav, O.; Lidbury, B.A.; Lewis, D.P.; Fisher, P.R. An Isolated Complex V Inefficiency and Dysregulated Mitochondrial Function in Immortalized Lymphocytes from ME/CFS Patients. Int. J. Mol. Sci. 2020, 21, 1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeus, M.; Nijs, J.; McGregor, N.; Meeusen, R.; De Schutter, G.; Truijen, S.; Frémont, M.; Van Hoof, E.; De Meirleir, K. Unravelling intracellular immune dysfunctions in chronic fatigue syndrome: Interactions between protein kinase R activity, RNase L cleavage and elastase activity, and their clinical relevance. In Vivo 2008, 22, 115–121. [Google Scholar] [PubMed]
- Sweetman, E.; Noble, A.; Edgar, C.; Mackay, A.; Helliwell, A.; Vallings, R.; Ryan, M.; Tate, W. Current Research Provides Insight into the Biological Basis and Diagnostic Potential for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Diagnostics 2019, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Vernon, S.D.; Funk, S.; Bateman, L.; Stoddard, G.J.; Hammer, S.; Sullivan, K.; Bell, J.; Abbaszadeh, S.; Lipkin, W.I.; Komaroff, A.L. Orthostatic Challenge Causes Distinctive Symptomatic, Hemodynamic and Cognitive Responses in Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Med. (Lausanne) 2022, 9, 917019. [Google Scholar] [CrossRef]
- Sepúlveda, N.; Carneiro, J.; Lacerda, E.; Nacul, L. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome as a Hyper-Regulated Immune System Driven by an Interplay Between Regulatory T Cells and Chronic Human Herpesvirus Infections. Front. Immunol. 2019, 10, 2684. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [Green Version]
- Sweetman, E.; Ryan, M.; Edgar, C.; MacKay, A.; Vallings, R.; Tate, W. Changes in the transcriptome of circulating immune cells of a New Zealand cohort with myalgic encephalomyelitis/chronic fatigue syndrome. Int. J. Immunopathol. Pharmacol. 2019, 33, 402. [Google Scholar] [CrossRef]
- Sweetman, E.; Kleffmann, T.; Edgar, C.; de Lange, M.; Vallings, R.; Tate, W. A SWATH-MS analysis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome peripheral blood mononuclear cell proteomes reveals mitochondrial dysfunction. J. Transl. Med. 2020, 18, 365. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, A.M.; Sweetman, E.C.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Changes in DNA methylation profiles of myalgic encephalomyelitis/chronic fatigue syndrome patients reflect systemic dysfunctions. Clin. Epigenet. 2020, 12, 167. [Google Scholar] [CrossRef]
- Nakatomi, Y.; Mizuno, K.; Ishii, A.; Wada, Y.; Tanaka, M.; Tazawa, S.; Onoe, K.; Fukuda, S.; Kawabe, J.; Takahashi, K.; et al. Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An (1)(1)C-(R)-PK11195 PET Study. J. Nucl. Med. 2014, 55, 945–950. [Google Scholar] [CrossRef] [Green Version]
- Visser, D.; Golla, S.S.V.; Verfaillie, S.C.J.; Coomans, E.M.; Rikken, R.M.; van de Giessen, E.M.; den Hollander, M.E.; Verveen, A.; Yaqub, M.; Barkhof, F.; et al. Long COVID is associated with extensive in-vivoneuroinflammation on [18F]DPA-714 PET. medRxiv 2022. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- VanElzakker, M.B.; Brumfield, S.A.; Lara Mejia, P.S. Neuroinflammation and Cytokines in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Critical Review of Research Methods. Front. Neurol. 2018, 9, 1033. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Lin, J.C.; Sheriff, S.; Maudsley, A.A.; Younger, J.W. Evidence of widespread metabolite abnormalities in Myalgic encephalomyelitis/chronic fatigue syndrome: Assessment with whole-brain magnetic resonance spectroscopy. Brain Imaging Behav. 2020, 14, 562–572. [Google Scholar] [CrossRef]
- Nelson, T.; Zhang, L.X.; Guo, H.; Nacul, L.; Song, X. Brainstem Abnormalities in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Scoping Review and Evaluation of Magnetic Resonance Imaging Findings. Front. Neurol. 2021, 12, 769511. [Google Scholar] [CrossRef]
- Quadt, L.; Critchley, H.D.; Garfinkel, S.N. The neurobiology of interoception in health and disease. Ann. N. Y. Acad. Sci. 2018, 1428, 112–128. [Google Scholar] [CrossRef]
- Schreiner, P.; Harrer, T.; Scheibenbogen, C.; Lamer, S.; Schlosser, A.; Naviaux, R.K.; Prusty, B.K. Human Herpesvirus-6 Reactivation, Mitochondrial Fragmentation, and the Coordination of Antiviral and Metabolic Phenotypes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Immunohorizons 2020, 4, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Bested, A.C.; Saunders, P.R.; Logan, A.C. Chronic fatigue syndrome: Neurological findings may be related to blood–brain barrier permeability. Med. Hypotheses 2001, 57, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, Y.; Chen, Z.; Leng, S.X. Connection between Systemic Inflammation and Neuroinflammation Underlies Neuroprotective Mechanism of Several Phytochemicals in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 1972714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olofsson, P.S.; Rosas-Ballina, M.; Levine, Y.A.; Tracey, K.J. Rethinking inflammation: Neural circuits in the regulation of immunity. Immunol. Rev. 2012, 248, 188–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, L.R.; Maier, S.F. Implications of immune-to-brain communication for sickness and pain. Proc. Natl. Acad. Sci. USA 1999, 96, 7710–7713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Thau, L.; Gandhi, J.; Sharma, S. Physiology, Cortisol. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Morris, G.; Anderson, G.; Maes, M. Hypothalamic-Pituitary-Adrenal Hypofunction in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a Consequence of Activated Immune-Inflammatory and Oxidative and Nitrosative Pathways. Mol. Neurobiol. 2017, 54, 6806–6819. [Google Scholar] [CrossRef]
- Hannibal, K.E.; Bishop, M.D. Chronic stress, cortisol dysfunction, and pain: A psychoneuroendocrine rationale for stress management in pain rehabilitation. Phys. Ther. 2014, 94, 1816–1825. [Google Scholar] [CrossRef]
- Straub, R.H.; Cutolo, M. Glucocorticoids and chronic inflammation. Rheumatology (Oxford) 2016, 55, ii6–ii14. [Google Scholar] [CrossRef] [Green Version]
- Pereira, G.; Gillies, H.; Chanda, S.; Corbett, M.; Vernon, S.D.; Milani, T.; Bateman, L. Acute Corticotropin-Releasing Factor Receptor Type 2 Agonism Results in Sustained Symptom Improvement in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Syst. Neurosci. 2021, 15, 698240. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Huenchuguala, S. Aminochrome Induces Irreversible Mitochondrial Dysfunction by Inducing Autophagy Dysfunction in Parkinson’s Disease. Front. Neurosci. 2018, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinan, T.G. Serotonin and the regulation of hypothalamic-pituitary-adrenal axis function. Life Sci. 1996, 58, 1683–1694. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back! Biomolecules 2021, 11, 185. [Google Scholar] [CrossRef]
- Williams, M.V.; Cox, B.; Lafuse, W.P.; Ariza, M.E. Epstein-Barr Virus dUTPase Induces Neuroinflammatory Mediators: Implications for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Clin. Ther. 2019, 41, 848–863. [Google Scholar] [CrossRef] [Green Version]
- Aïd, S.; Bosetti, F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasimir, F.; Toomey, D.; Liu, Z.; Kaiping, A.C.; Ariza, M.E.; Prusty, B.K. Tissue specific signature of HHV-6 infection in ME/CFS. Front. Mol. Biosci. 2022, 9, 1044964. [Google Scholar] [CrossRef]
- Hillman, E.T.; Lu, H.; Yao, T.; Nakatsu, C.H. Microbial Ecology along the Gastrointestinal Tract. Microbes Environ. 2017, 32, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, M.; Cao, J.; Li, X.; Fan, D.; Xia, Y.; Lu, X.; Li, J.; Ju, D.; Zhao, H. The Dynamic Interplay between the Gut Microbiota and Autoimmune Diseases. J. Immunol. Res. 2019, 2019, 7546047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giloteaux, L.; Goodrich, J.K.; Walters, W.A.; Levine, S.M.; Ley, R.E.; Hanson, M.R. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2016, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Newberry, F.; Hsieh, S.Y.; Wileman, T.; Carding, S.R. Does the microbiome and virome contribute to myalgic encephalomyelitis/chronic fatigue syndrome? Clin. Sci. (Lond.) 2018, 132, 523–542. [Google Scholar] [CrossRef] [Green Version]
- Wallis, A.; Ball, M.; Butt, H.; Lewis, D.P.; McKechnie, S.; Paull, P.; Jaa-Kwee, A.; Bruck, D. Open-label pilot for treatment targeting gut dysbiosis in myalgic encephalomyelitis/chronic fatigue syndrome: Neuropsychological symptoms and sex comparisons. J. Transl. Med. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, A.V.; Bested, A.C.; Beaulne, T.M.; Katzman, M.A.; Iorio, C.; Berardi, J.M.; Logan, A.C. A randomized, double-blind, placebo-controlled pilot study of a probiotic in emotional symptoms of chronic fatigue syndrome. Gut Pathog. 2009, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, A.; Nord, C.E.; Evengård, B. Effect of supplement with lactic-acid producing bacteria on fatigue and physical activity in patients with chronic fatigue syndrome. Nutr. J. 2009, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Borody, T.J.; Nowak, A.; Finlayson, S. The GI microbiome and its role in Chronic Fatigue Syndrome: A summary of bacteriotherapy. J. Australas. Coll. Nutr. Environ. Med. 2012, 31, 3–8. [Google Scholar]
- Guo, C.; Che, X.; Briese, T.; Ranjan, A.; Allicock, O.; Yates, R.A.; Cheng, A.; March, D.; Hornig, M.; Komaroff, A.L.; et al. Deficient butyrate-producing capacity in the gut microbiome is associated with bacterial network disturbances and fatigue symptoms in ME/CFS. Cell Host Microbe 2023, 31, 288–304. [Google Scholar] [CrossRef]
- Xiong, R.; Gunter, C.; Fleming, E.; Vernon, S.D.; Bateman, L.; Unutmaz, D.; Oh, J. Multi-’omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients. Cell Host Microbe 2023, 31, 273–287. [Google Scholar] [CrossRef]
- Hanson, M.R.; Giloteaux, L. The gut microbiome in Myalgic Encephalomyelitis. Biochemist 2017, 39, 10–13. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Lin, T.L.; Chang, C.J.; Wu, T.R.; Lai, W.F.; Lu, C.C.; Lai, H.C. Probiotics, prebiotics and amelioration of diseases. J. Biomed. Sci. 2019, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Cook, D.; Meyer, J.; Vernon, S.D.; Le, T.; Clevidence, D.; Robertson, C.E.; Schrodi, S.J.; Yale, S.; Frank, D.N. Changes in Gut and Plasma Microbiome following Exercise Challenge in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). PLoS ONE 2015, 10, e0145453. [Google Scholar] [CrossRef]
- Maes, M.; Kubera, M.; Leunis, J.C.; Berk, M. Increased IgA and IgM responses against gut commensals in chronic depression: Further evidence for increased bacterial translocation or leaky gut. J. Affect. Disord. 2012, 141, 55–62. [Google Scholar] [CrossRef]
- Sheedy, J.R.; Wettenhall, R.E.; Scanlon, D.; Gooley, P.R.; Lewis, D.P.; McGregor, N.; Stapleton, D.I.; Butt, H.L.; KL, D.E.M. Increased d-lactic Acid intestinal bacteria in patients with chronic fatigue syndrome. In Vivo 2009, 23, 621–628. [Google Scholar]
- Bested, A.C.; Logan, A.C.; Selhub, E.M. Intestinal microbiota, probiotics and mental health: From Metchnikoff to modern advances: Part I—Autointoxication revisited. Gut Pathog. 2013, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, J.N.; Coe, S.; Izadi, H. A retrospective outcome study of 42 patients with Chronic Fatigue Syndrome, 30 of whom had Irritable Bowel Syndrome. Half were treated with oral approaches, and half were treated with Faecal Microbiome Transplantation. Hum. Microbiome J. 2019, 13, 100061. [Google Scholar] [CrossRef]
- König, R.S.; Albrich, W.C.; Kahlert, C.R.; Bahr, L.S.; Löber, U.; Vernazza, P.; Scheibenbogen, C.; Forslund, S.K. The Gut Microbiome in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS). Front. Immunol. 2021, 12, 628741. [Google Scholar] [CrossRef] [PubMed]
- Castro-Marrero, J.; Domingo, J.C.; Cordobilla, B.; Ferrer, R.; Giralt, M.; Sanmartín-Sentañes, R.; Alegre-Martín, J. Does Coenzyme Q10 Plus Selenium Supplementation Ameliorate Clinical Outcomes by Modulating Oxidative Stress and Inflammation in Individuals with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome? Antioxid. Redox Signal. 2022, 36, 729–739. [Google Scholar] [CrossRef]
- Barletta, M.A.; Marino, G.; Spagnolo, B.; Bianchi, F.P.; Falappone, P.C.F.; Spagnolo, L.; Gatti, P. Coenzyme Q10 + alpha lipoic acid for chronic COVID syndrome. Clin. Exp. Med. 2022, 22, 1–12. [Google Scholar] [CrossRef]
- McInnes, I.B.; Gravallese, E.M. Immune-mediated inflammatory disease therapeutics: Past, present and future. Nat. Rev. Immunol. 2021, 21, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Fluge, Ø.; Bruland, O.; Risa, K.; Storstein, A.; Kristoffersen, E.K.; Sapkota, D.; Næss, H.; Dahl, O.; Nyland, H.; Mella, O. Benefit from B-lymphocyte depletion using the anti-CD20 antibody rituximab in chronic fatigue syndrome. A double-blind and placebo-controlled study. PLoS ONE 2011, 6, e26358. [Google Scholar] [CrossRef] [Green Version]
- Fluge, Ø.; Risa, K.; Lunde, S.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Kristoffersen, E.K.; Sørland, K.; Bruland, O.; Dahl, O.; et al. B-Lymphocyte Depletion in Myalgic Encephalopathy/Chronic Fatigue Syndrome. An Open-Label Phase II Study with Rituximab Maintenance Treatment. PLoS ONE 2015, 10, e0129898. [Google Scholar] [CrossRef] [PubMed]
- Fluge, Ø.; Rekeland, I.G.; Lien, K.; Thürmer, H.; Borchgrevink, P.C.; Schäfer, C.; Sørland, K.; Aßmus, J.; Ktoridou-Valen, I.; Herder, I.; et al. B-Lymphocyte Depletion in Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Ann. Intern. Med. 2019, 170, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.J.; Chapman, B.P.; Van Marwijk, H. Low-dose naltrexone as a treatment for chronic fatigue syndrome. BMJ Case Rep. 2020, 13, e232502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, O.; Pesonen, P.; Tuominen, E. Low-dose naltrexone in the treatment of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Fatigue Biomed. Health Behav. 2019, 7, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Eaton-Fitch, N.; Du Preez, S.; Cabanas, H.; Muraki, K.; Staines, D.; Marshall-Gradisnik, S. Impaired TRPM3-dependent calcium influx and restoration using Naltrexone in natural killer cells of myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Transl. Med. 2022, 20, 94. [Google Scholar] [CrossRef]
- Volpi-Abadie, J.; Kaye, A.M.; Kaye, A.D. Serotonin syndrome. Ochsner J. 2013, 13, 533–540. [Google Scholar]
- Kashi, A.A.; Davis, R.W.; Phair, R.D. The IDO Metabolic Trap Hypothesis for the Etiology of ME/CFS. Diagnostics 2019, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Lemle, M.D.; Komaroff, A.L.; Snyder, S.H. Redox imbalance links COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2021, 118, 2024358118. [Google Scholar] [CrossRef]
- Gueven, N.; Ravishankar, P.; Eri, R.; Rybalka, E. Idebenone: When an antioxidant is not an antioxidant. Redox Biol. 2021, 38, 101812. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, A.V.; Shchukin, I.A.; Soldatov, M.A.; Boĭko, O.V.; Petrov, S.V.; Khozova, A.A.; Ismailov, A.M.; Shikhkerimov, R.K.; Boĭko, A.N. [Asthenia, emotional disorders and quality of life of patients with multiple sclerosis]. Zhurnal Nevrol. I Psikhiatrii Im. SS Korsakova 2014, 114, 99–104. [Google Scholar]
- Tereshin, A.E.; Kiryanova, V.V.; Reshetnik, D.A. Correction of Mitochondrial Dysfunction in the Complex Rehabilitation of COVID-19 Patients. Neurosci. Behav. Physiol. 2022, 52, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.F.; Nilsson, M.; Eriksson, P.S.; Sims, N.R. Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci. Lett. 2004, 354, 163–165. [Google Scholar] [CrossRef]
- Li, J.; Sapper, T.N.; Mah, E.; Rudraiah, S.; Schill, K.E.; Chitchumroonchokchai, C.; Moller, M.V.; McDonald, J.D.; Rohrer, P.R.; Manautou, J.E.; et al. Green tea extract provides extensive Nrf2-independent protection against lipid accumulation and NFκB pro- inflammatory responses during nonalcoholic steatohepatitis in mice fed a high-fat diet. Mol. Nutr. Food Res. 2016, 60, 858–870. [Google Scholar] [CrossRef] [Green Version]
- Frei, B.; England, L.; Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 6377–6381. [Google Scholar] [CrossRef] [Green Version]
- Bruno, R.S.; Leonard, S.W.; Atkinson, J.; Montine, T.J.; Ramakrishnan, R.; Bray, T.M.; Traber, M.G. Faster plasma vitamin E disappearance in smokers is normalized by vitamin C supplementation. Free Radic. Biol. Med. 2006, 40, 689–697. [Google Scholar] [CrossRef]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 215–221. [Google Scholar] [CrossRef]
- Pretz, D.; Heyward, P.M.; Krebs, J.; Gruchot, J.; Barter, C.; Silcock, P.; Downes, N.; Rizwan, M.Z.; Boucsein, A.; Bender, J.; et al. A dahlia flower extract improves glucose homeostasis by improving insulin function in the brain. Medxiv 2023. in submission. [Google Scholar]
- White, P.D.; Goldsmith, K.A.; Johnson, A.L.; Potts, L.; Walwyn, R.; DeCesare, J.C.; Baber, H.L.; Burgess, M.; Clark, L.V.; Cox, D.L.; et al. Comparison of adaptive pacing therapy, cognitive behaviour therapy, graded exercise therapy, and specialist medical care for chronic fatigue syndrome (PACE): A randomised trial. Lancet 2011, 377, 823–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, M.; Goldsmith, K.A.; Johnson, A.L.; Chalder, T.; Walker, J.; White, P.D. Rehabilitative treatments for chronic fatigue syndrome: Long-term follow-up from the PACE trial. Lancet Psychiatry 2015, 2, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- NICE. Myalgic Encephalomyelitis (or Encephalopathy)/Chronic Fatigue Syndrome: Diagnosis and Management. Available online: https://www.nice.org.uk/guidance/ng206 (accessed on 22 January 2023).
- ANZMES. Lightning Process. Available online: https://anzmes.org.nz/lightning-process-switch/ (accessed on 22 January 2023).
- Hopper, A. Wired for Healing; Friesens: Altona, MB, Canada, 2014. [Google Scholar]
- Reesor, C. A Nurse with ME/CFS Finds Help in a Surprising Place: Christine’s DNRS Recovery Story. 2020. Available online: https://www.healthrising.org/blog/2020/01/05/christines-dnrs-chronic-fatigue-recovery-story/ (accessed on 22 January 2022).
- Bateman, L.; Bested, A.C.; Bonilla, H.F.; Chheda, B.V.; Chu, L.; Curtin, J.M.; Dempsey, T.T.; Dimmock, M.E.; Dowell, T.G.; Felsenstein, D.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Essentials of Diagnosis and Management. Mayo Clin. Proc. 2021, 96, 2861–2878. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||
| Severity of COVID-19 Infection | Long COVID Participants, n (%) | ME/CFS Trigger | ME/CFS Participants, n (%) |
| ICU/Hospitalised | 5 (8.8) | Bacterial infection | 4 (2.5) |
| Bed-bound | 26 (45.6) | Viral infection | 86 (53.8) |
| House-bound | 16 (28.1) | Immune system problems | 2 (1.3) |
| Mild symptoms | 9 (15.8) | Emotional trauma | 7 (4.4) |
| No Symptoms | 1 (1.8) | Physical trauma | 6 (3.8) |
| Other | 55 (34.4) | ||
| Total | 57 (100) | Total | 160 (100) |
| (B) | |||
| Underlying Health Conditions | Long COVID Participants, n (%) | ME/CFS Participants, n (%) | Total, n (%) |
| Gastrointestinal issues | 19 (33.3) | 73 (45.6) | 92 (42.4) |
| Mental health condition | 10 (17.5) | 58 (36.3) | 68 (31.3) |
| Allergies | 9 (15.8) | 27 (16.9) | 36 (16.6) |
| Autoimmune condition | 8 (14.0) | 25 (15.6) | 33 (15.2) |
| Inflammatory disease | 9 (15.8) | 18 (11.3) | 27 (12.4) |
| Lifelong low energy | 2 (3.5) | 24 (15.0) | 26 (12.0) |
| Cancer | 0 (0.0) | 3 (1.9) | 3 (1.4) |
| Other | 30 (52.6) | 69 (43.1) | 99 (45.6) |
| None | 11 (19.3) | 25 (15.6) | 36 (16.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tate, W.P.; Walker, M.O.M.; Peppercorn, K.; Blair, A.L.H.; Edgar, C.D. Towards a Better Understanding of the Complexities of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID. Int. J. Mol. Sci. 2023, 24, 5124. https://doi.org/10.3390/ijms24065124
Tate WP, Walker MOM, Peppercorn K, Blair ALH, Edgar CD. Towards a Better Understanding of the Complexities of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID. International Journal of Molecular Sciences. 2023; 24(6):5124. https://doi.org/10.3390/ijms24065124
Chicago/Turabian StyleTate, Warren P., Max O. M. Walker, Katie Peppercorn, Anna L. H. Blair, and Christina D. Edgar. 2023. "Towards a Better Understanding of the Complexities of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID" International Journal of Molecular Sciences 24, no. 6: 5124. https://doi.org/10.3390/ijms24065124
APA StyleTate, W. P., Walker, M. O. M., Peppercorn, K., Blair, A. L. H., & Edgar, C. D. (2023). Towards a Better Understanding of the Complexities of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID. International Journal of Molecular Sciences, 24(6), 5124. https://doi.org/10.3390/ijms24065124