

Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
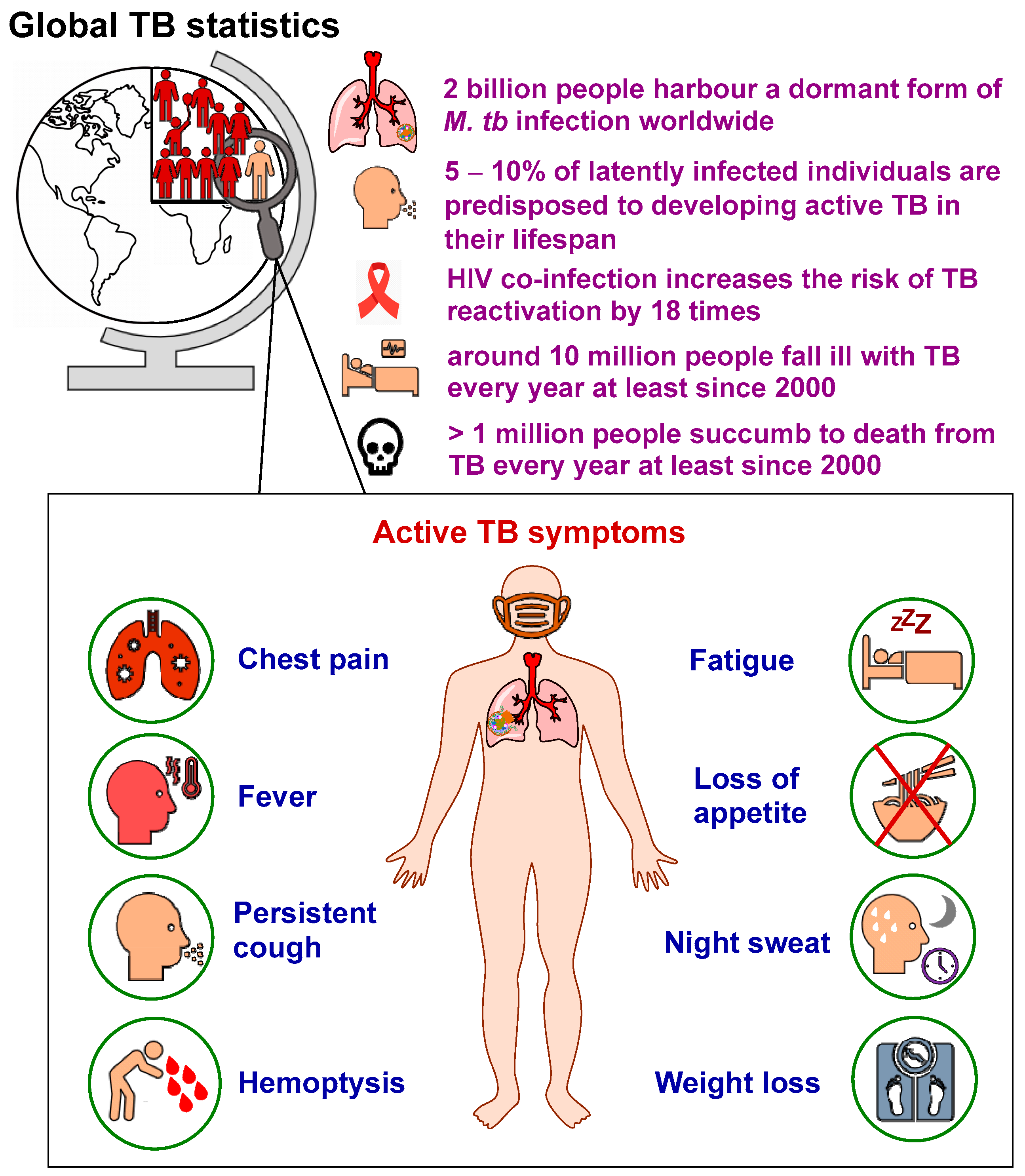
:1. Introduction
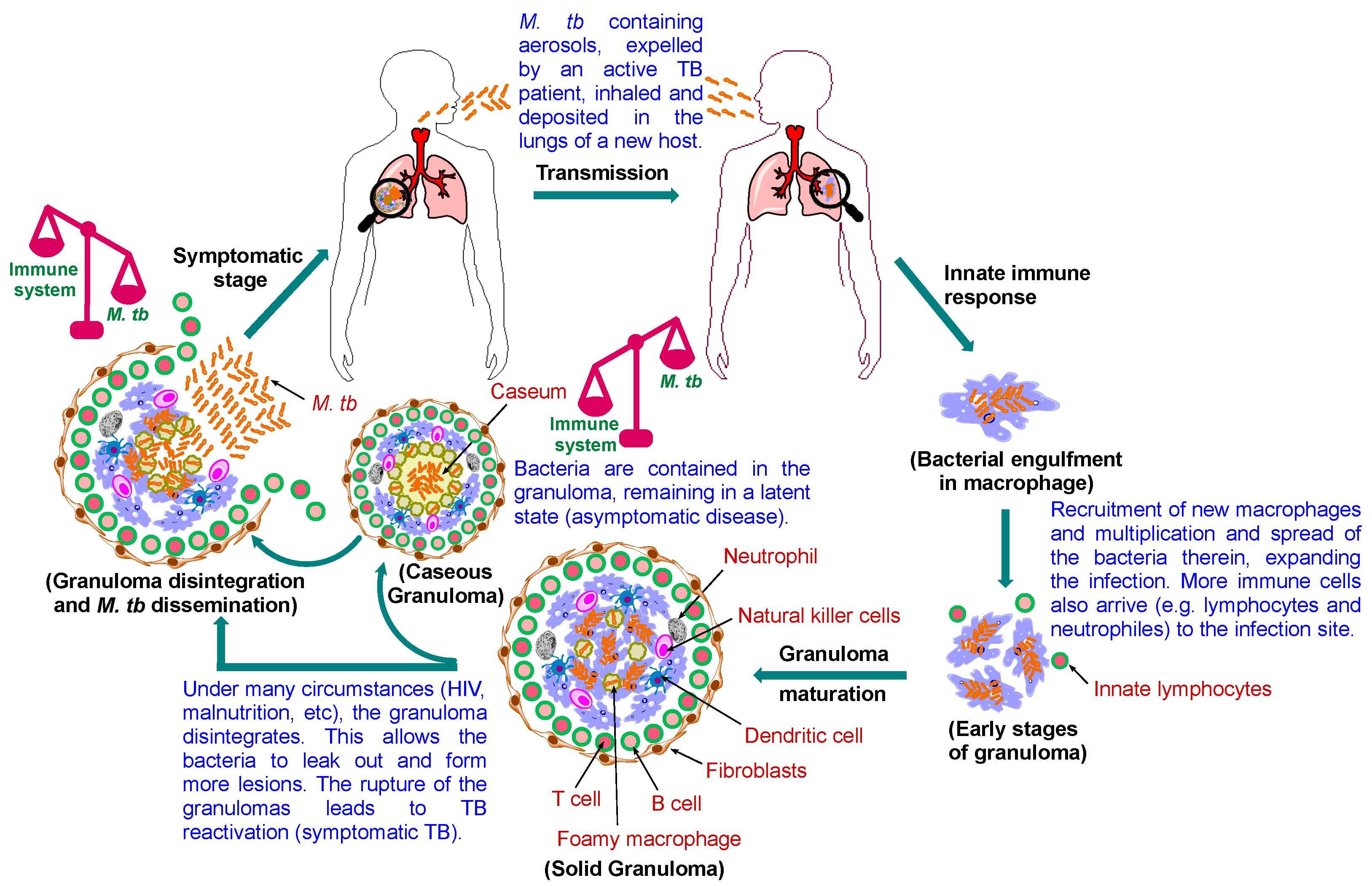
2. TB Pathogenesis
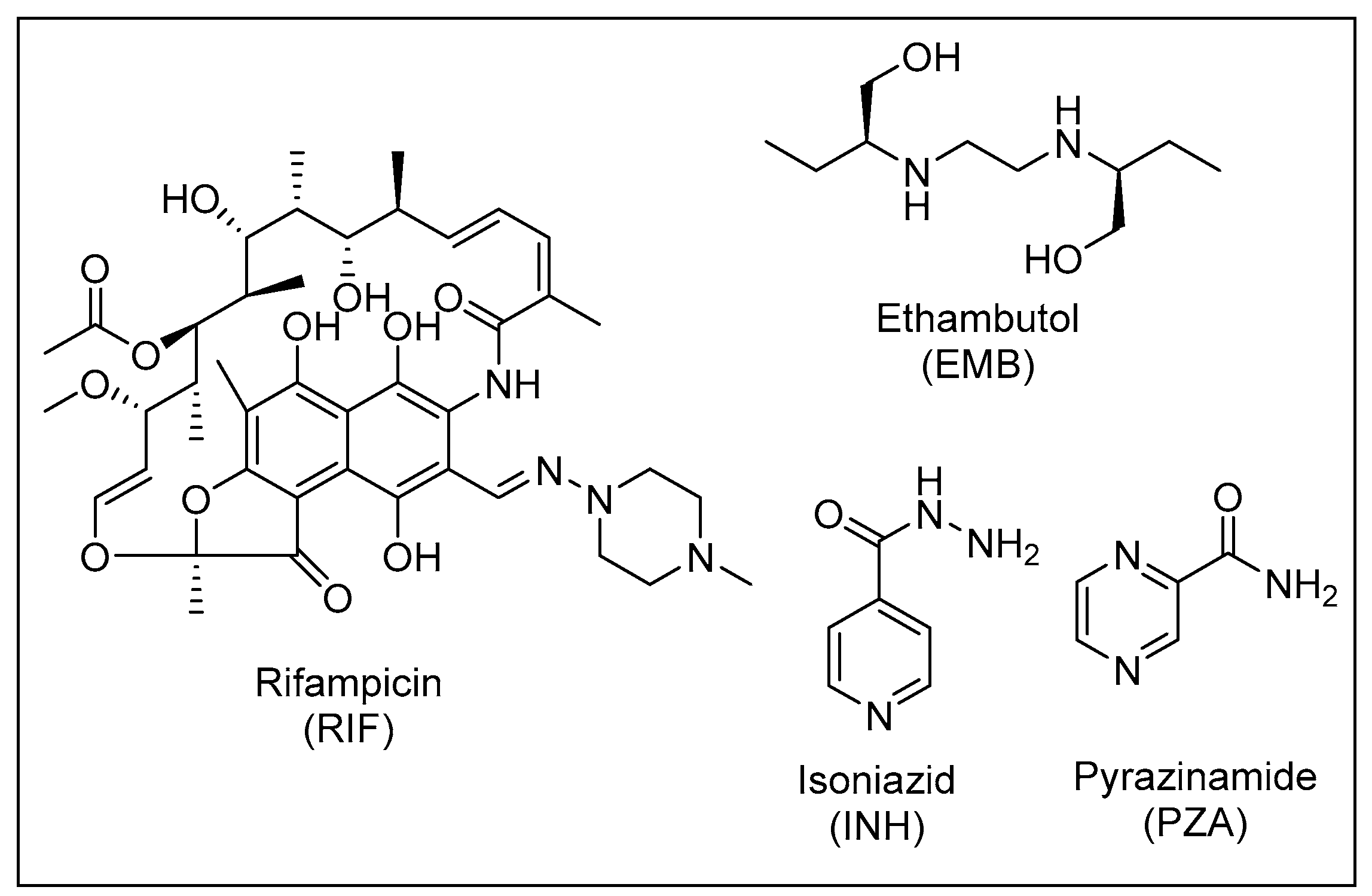
3. Current Treatment Regimen for Drug-Sensitive (DS) TB
4. Challenges to the Global Control of TB
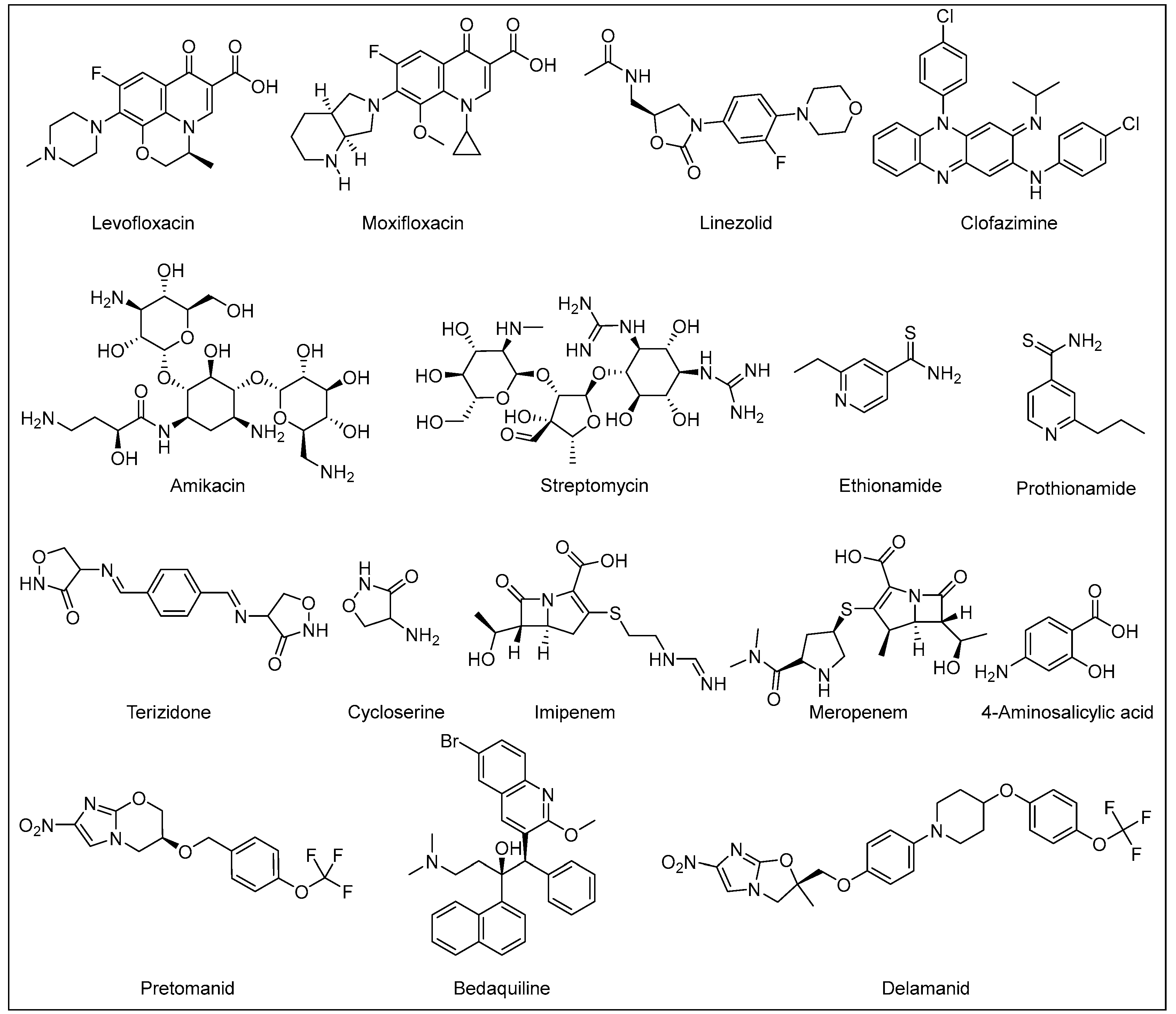
4.1. Drug-Resistant (DR) TB Crisis
4.2. TB and HIV Co-Infection
4.3. The Coronavirus 2019 (COVID-19) Pandemic and TB
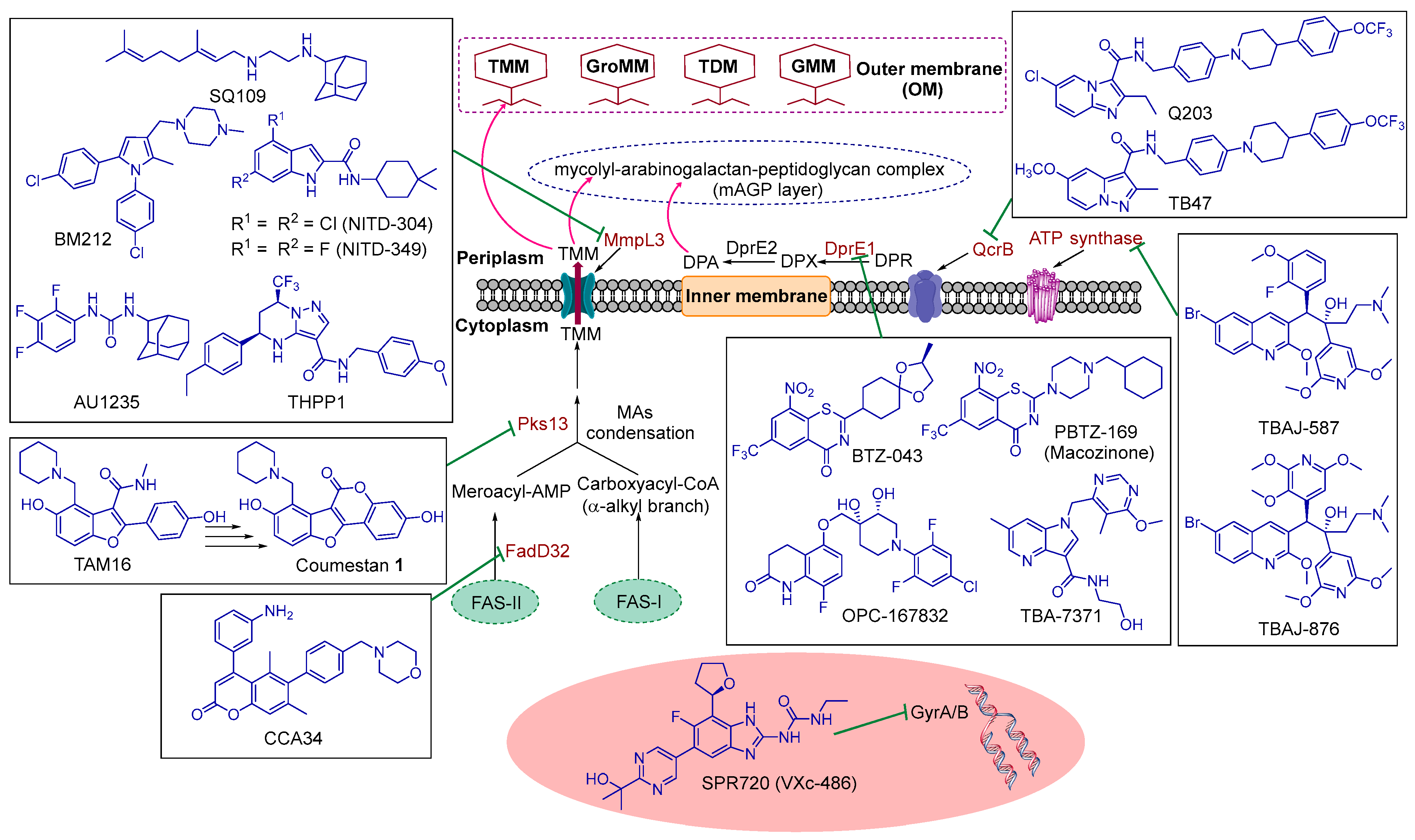
5. TB Drug Targets
5.1. Overview
5.2. Current Hot Targets in M. tb Drug Discovery and Their Corresponding TB Drug Candidates
5.2.1. GyrA/B
5.2.2. ATP Synthase
5.2.3. QcrB
5.2.4. DprE1
5.2.5. FadD32 and Pks13
5.2.6. MmpL3
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaufmann, S.H.; Schaible, U.E. 100th anniversary of Robert Koch’s Nobel Prize for the discovery of the tubercle bacillus. Trends Microbiol. 2005, 13, 469–475. [Google Scholar] [CrossRef]
- World Health Organisation. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022; Licence: CC BY-NC-SA 3.0 IGO; Available online: https://www.who.int/publications/i/item/9789240061729 (accessed on 1 February 2023).
- Flynn, J.L.; Chan, J. Tuberculosis: Latency and Reactivation. Infect. Immun. 2001, 69, 4195–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organisation. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; Licence: CC BY-NC-SA 3.0 IGO; Available online: https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf (accessed on 1 February 2023).
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.; Acharya, A.; Gautam, S.; Ghimire, S.P.; Mishra, G.; Parajuli, N.; Sapkota, B. Advances in diagnosis of Tuberculosis: An update into molecular diagnosis of Mycobacterium tuberculosis. Mol. Biol. Rep. 2020, 47, 4065–4075. [Google Scholar] [CrossRef]
- Leung, A.N. Pulmonary Tuberculosis: The Essentials. Radiology 1999, 210, 307–322. [Google Scholar] [CrossRef]
- Luies, L.; du Preez, I. The Echo of Pulmonary Tuberculosis: Mechanisms of Clinical Symptoms and Other Disease-Induced Systemic Complications. Clin. Microbiol. Rev. 2020, 33, e00036-20. [Google Scholar] [CrossRef] [PubMed]
- Schluger, N.W. The pathogenesis of tuberculosis: The first one hundred (and twenty-three) years. Am. J. Respir. Cell Mol. Biol. 2005, 32, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Cardona, P.-J.; Kim, M.-J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huszár, S.; Chibale, K.; Singh, V. The quest for the holy grail: New antitubercular chemical entities, targets and strategies. Drug Discov. Today 2020, 25, 772–780. [Google Scholar] [CrossRef]
- Chai, Q.; Zhang, Y.; Liu, C.H. Mycobacterium tuberculosis: An Adaptable Pathogen Associated With Multiple Human Diseases. Front. Cell. Infect. Microbiol. 2018, 8, 158. [Google Scholar] [CrossRef]
- Marrakchi, H.; Laneelle, M.A.; Daffe, M. Mycolic acids: Structures, biosynthesis, and beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosset, J. Mycobacterium tuberculosis in the Extracellular Compartment: An Underestimated Adversary. Antimicrob. Agents Chemother. 2003, 47, 833–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorhoi, A.; Kaufmann, S.H. Pathology and immune reactivity: Understanding multidimensionality in pulmonary tuberculosis. Semin. Immunopathol. 2015, 38, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.L. Pathology of post primary tuberculosis of the lung: An illustrated critical review. Tuberculosis 2011, 91, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reece, S.T.; Kaufmann, S.H. Floating between the poles of pathology and protection: Can we pin down the granuloma in tuberculosis? Curr. Opin. Microbiol. 2012, 15, 63–70. [Google Scholar] [CrossRef]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Louw, G.E.; Warren, R.M.; van Pittius, N.C.G.; Leon, R.; Jimenez, A.; Hernandez-Pando, R.; McEvoy, C.R.E.; Grobbelaar, M.; Murray, M.; van Helden, P.D.; et al. Rifampicin Reduces Susceptibility to Ofloxacin in Rifampicin-resistant Mycobacterium tuberculosis through Efflux. Am. J. Respir. Crit. Care Med. 2011, 184, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar] [PubMed]
- Goude, R.; Amin, A.G.; Chatterjee, D.; Parish, T. The Arabinosyltransferase EmbC Is Inhibited by Ethambutol in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 4138–4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.-Y.; Centis, R.; Migliori, G.B. Drug-resistant tuberculosis: Past, present, future. Respirology 2010, 15, 413–432. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Centis, R.; D’Ambrosio, L.; Migliori, G.B. Tuberculosis Treatment and Drug Regimens. Cold Spring Harb. Perspect. Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [Green Version]
- Yee, D.; Valiquette, C.; Pelletier, M.; Parisien, I.; Rocher, I.; Menzies, D. Incidence of Serious Side Effects from First-Line Antituberculosis Drugs among Patients Treated for Active Tuberculosis. Am. J. Respir. Crit. Care Med. 2003, 167, 1472–1477. [Google Scholar] [CrossRef]
- World Health Organisation. WHO Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2019; Licence: CC BY-NC-SA 3.0 IGO; Available online: https://apps.who.int/iris/bitstream/handle/10665/311389/9789241550529-eng.pdf (accessed on 1 February 2023).
- Migliori, G.B.; Tiberi, S.; Zumla, A.; Petersen, E.; Chakaya, J.M.; Wejse, C.; Muñoz Torrico, M.; Duarte, R.; Alffenaar, J.W.; Schaaf, H.S.; et al. MDR/XDR-TB management of patients and contacts: Challenges facing the new decade. The 2020 clinical update by the Global Tuberculosis Network. Int. J. Infect. Dis. 2020, 92S, S15–S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organisation. Module 4: Treatment—Drug-Resistant Tuberculosis Treatment. In WHO Operational Handbook on Tuberculosis; World Health Organization: Geneva, Switzerland, 2020; Licence: CC BY-NC-SA 3.0 IGO; Available online: https://www.who.int/publications/i/item/9789240006997 (accessed on 1 February 2023).
- Zumla, A.; Chakaya, J.; Centis, R.; D’Ambrosio, L.; Mwaba, P.; Bates, M.; Kapata, N.; Nyirenda, T.; Chanda, D.; Mfinanga, S.; et al. Tuberculosis treatment and management--an update on treatment regimens, trials, new drugs, and adjunct therapies. Lancet Respir. Med. 2015, 3, 220–234. [Google Scholar] [CrossRef]
- Sensi, P. History of the Development of Rifampin. Clin. Infect. Dis. 1983, 5 (Suppl. S3), S402–S406. [Google Scholar] [CrossRef]
- Nahid, P.; Mase, S.R.; Migliori, G.B.; Sotgiu, G.; Bothamley, G.H.; Brozek, J.L.; Cattamanchi, A.; Cegielski, J.P.; Chen, L.; Daley, C.L.; et al. Treatment of Drug-Resistant Tuberculosis. An Official ATS/CDC/ERS/IDSA Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2019, 200, e93–e142. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Mudde, S.E.; Upton, A.M.; Lenaerts, A.; Bax, H.I.; De Steenwinkel, J.E.M. Delamanid or pretomanid? A Solomonic judgement! J. Antimicrob. Chemother. 2022, 77, 880–902. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, A.M.; Spigelman, M. Challenges in tuberculosis drug research and development. Nat. Med. 2007, 13, 290–294. [Google Scholar] [CrossRef]
- Pawlowski, A.; Jansson, M.; Skold, M.; Rottenberg, M.E.; Kallenius, G. Tuberculosis and HIV co-infection. PLoS Pathog. 2012, 8, e1002464. [Google Scholar] [CrossRef]
- World Health Organisation. COVID-19 Weekly Epidemiological Update on COVID-19. 25 January 2023. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---25-january-2023 (accessed on 1 February 2023).
- World Health Organisation. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; Licence: CC BY-NC-SA 3.0 IGO; Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 1 February 2023).
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mdluli, K.; Kaneko, T.; Upton, A. The Tuberculosis Drug Discovery and Development Pipeline and Emerging Drug Targets. Cold Spring Harb. Perspect. Med. 2015, 5, a021154. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, V.; Poce, G.; Canseco, J.O.; Buroni, S.; Pasca, M.R.; Biava, M.; Raju, R.M.; Porretta, G.C.; Alfonso, S.; Battilocchio, C.; et al. MmpL3 Is the Cellular Target of the Antitubercular Pyrrole Derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Manjunatha, U.H.; Smith, P.W. Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorganic Med. Chem. 2015, 23, 5087–5097. [Google Scholar] [CrossRef] [Green Version]
- Locher, C.P.; Jones, S.M.; Hanzelka, B.L.; Perola, E.; Shoen, C.M.; Cynamon, M.H.; Ngwane, A.H.; Wiid, I.J.; van Helden, P.D.; Betoudji, F.; et al. A Novel Inhibitor of Gyrase B Is a Potent Drug Candidate for Treatment of Tuberculosis and Nontuberculosis Mycobacterial Infections. Antimicrob. Agents Chemother. 2015, 59, 1455–1465. [Google Scholar] [CrossRef] [Green Version]
- Mdluli, K.; Ma, Z. Mycobacterium tuberculosis DNA gyrase as a target for drug discovery. Infect. Disord. Drug Targets 2007, 7, 159–168. [Google Scholar] [CrossRef]
- Bruch, E.M.; Petrella, S.; Bellinzoni, M. Structure-Based Drug Design for Tuberculosis: Challenges Still Ahead. Appl. Sci. 2020, 10, 4248. [Google Scholar] [CrossRef]
- 2022 Global New TB Drug Pipeline. Available online: https://www.newtbdrugs.org/pipeline/clinical (accessed on 1 February 2023).
- 2022 Global New TB Drug Pipeline. Available online: https://www.newtbdrugs.org/pipeline/discovery (accessed on 1 February 2023).
- Spero Therapeutics Announces Positive Results from SPR720 IND-Enabling Studies and Plans to Initiate a Phase 1 Trial. Available online: https://www.globenewswire.com/news-release/2018/11/05/1644995/0/en/Spero-Therapeutics-Announces-Positive-Results-from-SPR720-IND-Enabling-Studies-and-Plans-to-Initiate-a-Phase-1-Trial.html (accessed on 1 February 2023).
- Stout, J.E.; Koh, W.-J.; Yew, W.W. Update on pulmonary disease due to non-tuberculous mycobacteria. Int. J. Infect. Dis. 2016, 45, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spero Therapeutics Announces Initiation of SPR720 Phase 1 Clinical Trial. Available online: https://www.globenewswire.com/en/news-release/2019/01/29/1706794/0/en/Spero-Therapeutics-Announces-Initiation-of-SPR720-Phase-1-Clinical-Trial.html (accessed on 1 February 2023).
- Spero Therapeutics Receives QIDP Designation from the U.S. FDA for the Development of SPR720. Available online: https://www.globenewswire.com/news-release/2019/02/26/1742382/0/en/Spero-Therapeutics-Receives-QIDP-Designation-from-the-U-S-FDA-for-the-Development-of-SPR720.html (accessed on 1 February 2023).
- Spero Therapeutics Provides Update on SPR720 Phase 2a Clinical Trial. Available online: https://www.globenewswire.com/news-release/2021/02/05/2170670/0/en/Spero-Therapeutics-Provides-Update-on-SPR720-Phase-2a-Clinical-Trial.html (accessed on 1 February 2023).
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Lill, H.; Bald, D. ATP synthase in mycobacteria: Special features and implications for a function as drug target. Biochim. Biophys. Acta 2014, 1837, 1208–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, H.S.; Tong, A.S.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorganic Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Chahine, E.B.; Karaoui, L.R.; Mansour, H. Bedaquiline: A novel diarylquinoline for multidrug-resistant tuberculosis. Ann. Pharmacother. 2014, 48, 107–115. [Google Scholar] [CrossRef]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Lu, X.; Williams, Z.; Hards, K.; Tang, J.; Cheung, C.Y.; Aung, H.L.; Wang, B.; Liu, Z.; Hu, X.; Lenaerts, A.; et al. Pyrazolo [1,5-a]pyridine Inhibitor of the Respiratory Cytochrome bcc Complex for the Treatment of Drug-Resistant Tuberculosis. ACS Infect. Dis. 2019, 5, 239–249. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Barmade, M.A.; Murumkar, P.R.; Yadav, M.R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [Green Version]
- Brecik, M.; Centárová, I.; Mukherjee, R.; Kolly, G.S.; Huszár, S.; Bobovská, A.; Kilacsková, E.; Mokošová, V.; Svetlíková, Z.; Šarkan, M.; et al. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10, 1631–1636. [Google Scholar] [CrossRef]
- Kolly, G.S.; Boldrin, F.; Sala, C.; Dhar, N.; Hartkoorn, R.C.; Ventura, M.; Serafini, A.; McKinney, J.D.; Manganelli, R.; Cole, S.T. Assessing the essentiality of the decaprenyl-phospho-d-arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol. Microbiol. 2014, 92, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, C.; Skovierova, H.; Buroni, S.; Bobovska, A.; Nenci, S.; Molteni, E.; Pojer, F.; Pasca, M.R.; Makarov, V.; Cole, S.T.; et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-beta-D-ribofuranose 2’-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; Van Der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.; Ryabova, O.B.; Vocat, A.; Decosterd, L.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Neres, J.; Hartkoorn, R.C.; Ryabova, O.B.; Kazakova, E.; Šarkan, M.; Huszár, S.; Piton, J.; Kolly, G.S.; Vocat, A.; et al. The 8-Pyrrole-Benzothiazinones Are Noncovalent Inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4446–4452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariguchi, N.; Chen, X.; Hayashi, Y.; Kawano, Y.; Fujiwara, M.; Matsuba, M.; Shimizu, H.; Ohba, Y.; Nakamura, I.; Kitamoto, R.; et al. OPC-167832, a Novel Carbostyril Derivative with Potent Antituberculosis Activity as a DprE1 Inhibitor. Antimicrob. Agents Chemother. 2020, 64, e02020-19. [Google Scholar] [CrossRef] [Green Version]
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hameed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium tuberculosis and Are Efficacious in Vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [CrossRef]
- Shirude, P.S.; Shandil, R.K.; Manjunatha, M.R.; Sadler, C.; Panda, M.; Panduga, V.; Reddy, J.; Saralaya, R.; Nanduri, R.; Ambady, A.; et al. Lead Optimization of 1,4-Azaindoles as Antimycobacterial Agents. J. Med. Chem. 2014, 57, 5728–5737. [Google Scholar] [CrossRef]
- Chatterji, M.; Shandil, R.; Manjunatha, M.R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; Kr, P.; et al. 1,4-Azaindole, a Potential Drug Candidate for Treatment of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef] [Green Version]
- Early Bactericidal Activity of TBA-7371 in Pulmonary Tuberculosis. Available online: https://clinicaltrials.gov/ct2/show/NCT04176250 (accessed on 1 February 2023).
- Gavalda, S.; Bardou, F.; Laval, F.; Bon, C.; Malaga, W.; Chalut, C.; Guilhot, C.; Mourey, L.; Daffé, M.; Quémard, A. The Polyketide Synthase Pks13 Catalyzes a Novel Mechanism of Lipid Transfer in Mycobacteria. Chem. Biol. 2014, 21, 1660–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Gu, S.; Fleming, J.; Bi, L. Crystal structure of FadD32, an enzyme essential for mycolic acid biosynthesis in mycobacteria. Sci. Rep. 2015, 5, 15493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M.L.; Alexander, E.; Minasov, G.; Page, H.J.; Warwrzak, Z.; Shuvalova, L.; Flores, K.J.; Wilson, D.J.; Shi, C.; Aldrich, C.C.; et al. Structure of the Essential Mtb FadD32 Enzyme: A Promising Drug Target for Treating Tuberculosis. ACS Infect. Dis. 2016, 2, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavalda, S.; Leger, M.; van der Rest, B.; Stella, A.; Bardou, F.; Montrozier, H.; Chalut, C.; Burlet-Schiltz, O.; Marrakchi, H.; Daffe, M.; et al. The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J. Biol. Chem. 2009, 284, 19255–19264. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.; Molle, V.; Besra, G.S.; Jacobs, W.R., Jr.; Kremer, L. The Mycobacterium tuberculosis FAS-II condensing enzymes: Their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 2007, 64, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Portevin, D.; De Sousa-D’Auria, C.; Houssin, C.; Grimaldi, C.; Chami, M.; Daffe, M.; Guilhot, C. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. USA 2004, 101, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Portevin, D.; de Sousa-D’Auria, C.; Montrozier, H.; Houssin, C.; Stella, A.; Laneelle, M.A.; Bardou, F.; Guilhot, C.; Daffe, M. The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: Identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J. Biol. Chem. 2005, 280, 8862–8874. [Google Scholar]
- Gande, R.; Gibson, K.J.C.; Brown, A.K.; Krumbach, K.; Dover, L.; Sahm, H.; Shioyama, S.; Oikawa, T.; Besra, G.; Eggeling, L. Acyl-CoA Carboxylases (accD2 and accD3), Together with a Unique Polyketide Synthase (Cg-pks), Are Key to Mycolic Acid Biosynthesis in Corynebacterianeae Such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J. Biol. Chem. 2004, 279, 44847–44857. [Google Scholar] [CrossRef] [Green Version]
- Carroll, P.; Faray-Kele, M.-C.; Parish, T. Identifying Vulnerable Pathways in Mycobacterium tuberculosis by Using a Knockdown Approach. Appl. Environ. Microbiol. 2011, 77, 5040–5043. [Google Scholar] [CrossRef] [Green Version]
- Kawate, T.; Iwase, N.; Shimizu, M.; Stanley, S.A.; Wellington, S.; Kazyanskaya, E.; Hung, D.T. Synthesis and structure–activity relationships of phenyl-substituted coumarins with anti-tubercular activity that target FadD32. Bioorganic Med. Chem. Lett. 2013, 23, 6052–6059. [Google Scholar] [CrossRef]
- Stanley, S.A.; Kawate, T.; Iwase, N.; Shimizu, M.; Clatworthy, A.E.; Kazyanskaya, E.; Sacchettini, J.C.; Ioerger, T.R.; Siddiqi, N.A.; Minami, S.; et al. Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. USA 2013, 110, 11565–11570. [Google Scholar] [CrossRef] [Green Version]
- Ioerger, T.R.; O’Malley, T.; Liao, R.; Guinn, K.M.; Hickey, M.J.; Mohaideen, N.; Murphy, K.C.; Boshoff, H.I.; Mizrahi, V.; Rubin, E.J.; et al. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e75245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Lun, S.; Liu, L.L.; Xiao, S.; Duan, G.; Gunosewoyo, H.; Yang, F.; Tang, J.; Bishai, W.R.; Yu, L.F. Identification of Novel Coumestan Derivatives as Polyketide Synthase 13 Inhibitors against Mycobacterium tuberculosis. Part II. J. Med. Chem. 2019, 62, 3575–3589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lun, S.; Wang, S.H.; Jiang, X.W.; Yang, F.; Tang, J.; Manson, A.L.; Earl, A.M.; Gunosewoyo, H.; Bishai, W.R.; et al. Identification of Novel Coumestan Derivatives as Polyketide Synthase 13 Inhibitors against Mycobacterium tuberculosis. J. Med. Chem. 2018, 61, 791–803. [Google Scholar] [CrossRef]
- Lun, S.; Xiao, S.; Zhang, W.; Wang, S.; Gunosewoyo, H.; Yu, L.-F.; Bishai, W.R. Therapeutic Potential of Coumestan Pks13 Inhibitors for Tuberculosis. Antimicrob. Agents Chemother. 2021, 65. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.-S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef] [Green Version]
- Tima, H.G.; Al Dulayymi, J.R.; Denis, O.; Lehebel, P.; Baols, K.S.; Mohammed, M.O.; L’Homme, L.; Sahb, M.M.; Potemberg, G.; Legrand, S.; et al. Inflammatory Properties and Adjuvant Potential of Synthetic Glycolipids Homologous to Mycolate Esters of the Cell Wall of Mycobacterium tuberculosis. J. Innate Immun. 2016, 9, 162–180. [Google Scholar] [CrossRef]
- Degiacomi, G.; Benjak, A.; Madacki, J.; Boldrin, F.; Provvedi, R.; Palù, G.; Kordulakova, J.; Cole, S.T.; Manganelli, R. Essentiality of mmpL3 and impact of its silencing on Mycobacterium tuberculosis gene expression. Sci. Rep. 2017, 7, 43495. [Google Scholar] [CrossRef] [Green Version]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.; Kordulakova, J.; et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Obregón-Henao, A.; Wallach, J.B.; North, E.J.; Lee, R.E.; Gonzalez-Juarrero, M.; Schnappinger, D.; Jackson, M. Therapeutic Potential of the Mycobacterium tuberculosis Mycolic Acid Transporter, MmpL3. Antimicrob. Agents Chemother. 2016, 60, 5198–5207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, C.; Rittmann, D.; Singh, A.; Krumbach, K.; Bhatt, K.; Eggeling, L.; Besra, G.S.; Bhatt, A. MmpL Genes Are Associated with Mycolic Acid Metabolism in Mycobacteria and Corynebacteria. Chem. Biol. 2012, 19, 498–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.-C.; Klenotic, P.A.; Bolla, J.R.; Purdy, G.E.; Robinson, C.V.; Yu, E.W. MmpL3 is a lipid transporter that binds trehalose monomycolate and phosphatidylethanolamine. Proc. Natl. Acad. Sci. USA 2019, 116, 11241–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E., 3rd; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [Green Version]
- Lun, S.; Guo, H.; Onajole, O.K.; Pieroni, M.; Gunosewoyo, H.; Chen, G.; Tipparaju, S.K.; Ammerman, N.C.; Kozikowski, A.P.; Bishai, W.R. Indoleamides are active against drug-resistant Mycobacterium tuberculosis. Nat. Commun. 2013, 4, 2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.P.S.; Lakshminarayana, S.B.; Kondreddi, R.R.; Herve, M.; Camacho, L.R.; Bifani, P.; Kalapala, S.K.; Jiricek, J.; Ma, N.L.; Tan, B.H.; et al. Indolcarboxamide Is a Preclinical Candidate for Treating Multidrug-Resistant Tuberculosis. Sci. Transl. Med. 2013, 5, 214ra168. [Google Scholar] [CrossRef] [PubMed]
- Stec, J.; Onajole, O.K.; Lun, S.; Guo, H.; Merenbloom, B.; Vistoli, G.; Bishai, W.R.; Kozikowski, A.P. Indole-2-carboxamide-based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem. 2016, 59, 6232–6247. [Google Scholar] [CrossRef]
- Remuiñán, M.J.; Pérez-Herrán, E.; Rullás, J.; Alemparte, C.; Martínez-Hoyos, M.; Dow, D.J.; Afari, J.; Mehta, N.; Esquivias, J.; Jiménez, E.; et al. Tetrahydropyrazolo[1,5-a]Pyrimidine-3-Carboxamide and N-Benzyl-6′,7′-Dihydrospiro[Piperidine-4,4′-Thieno[3,2-c]Pyran] Analogues with Bactericidal Efficacy against Mycobacterium tuberculosis Targeting MmpL3. PLoS ONE 2013, 8, e60933. [Google Scholar] [CrossRef]
- Li, W.; Stevens, C.M.; Pandya, A.N.; Darzynkiewicz, Z.M.; Bhattarai, P.; Tong, W.; Gonzalez-Juarrero, M.; North, E.J.; Zgurskaya, H.I.; Jackson, M.C. Direct Inhibition of MmpL3 by Novel Antitubercular Compounds. ACS Infect. Dis. 2019, 5, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, J.; Yang, X.; Wu, L.; Zhang, J.; Yang, Y.; Zhao, Y.; Zhang, L.; Yang, X.; Yang, X.; et al. Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell 2019, 176, 636–648.e13. [Google Scholar] [CrossRef] [Green Version]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E., 3rd; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Deidda, D.; Lampis, G.; Fioravanti, R.; Biava, M.; Porretta, G.C.; Zanetti, S.; Pompei, R. Bactericidal Activities of the Pyrrole Derivative BM212 against Multidrug-Resistant and Intramacrophagic Mycobacterium tuberculosis Strains. Antimicrob. Agents Chemother. 1998, 42, 3035–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.R.; North, E.J.; Hurdle, J.G.; Morisseau, C.; Scarborough, J.S.; Sun, D.; Korduláková, J.; Scherman, M.S.; Jones, V.; Grzegorzewicz, A.; et al. The structure–activity relationship of urea derivatives as anti-tuberculosis agents. Bioorganic. Med. Chem. 2011, 19, 5585–5595. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int. J. Mol. Sci. 2023, 24, 5202. https://doi.org/10.3390/ijms24065202
Alsayed SSR, Gunosewoyo H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. International Journal of Molecular Sciences. 2023; 24(6):5202. https://doi.org/10.3390/ijms24065202
Chicago/Turabian StyleAlsayed, Shahinda S. R., and Hendra Gunosewoyo. 2023. "Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets" International Journal of Molecular Sciences 24, no. 6: 5202. https://doi.org/10.3390/ijms24065202
APA StyleAlsayed, S. S. R., & Gunosewoyo, H. (2023). Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. International Journal of Molecular Sciences, 24(6), 5202. https://doi.org/10.3390/ijms24065202