
Importance of Germline and Somatic Alterations in Human MRE11, RAD50, and NBN Genes Coding for MRN Complex

 , , , , , and
, , , , , and
Abstract
:1. Introduction
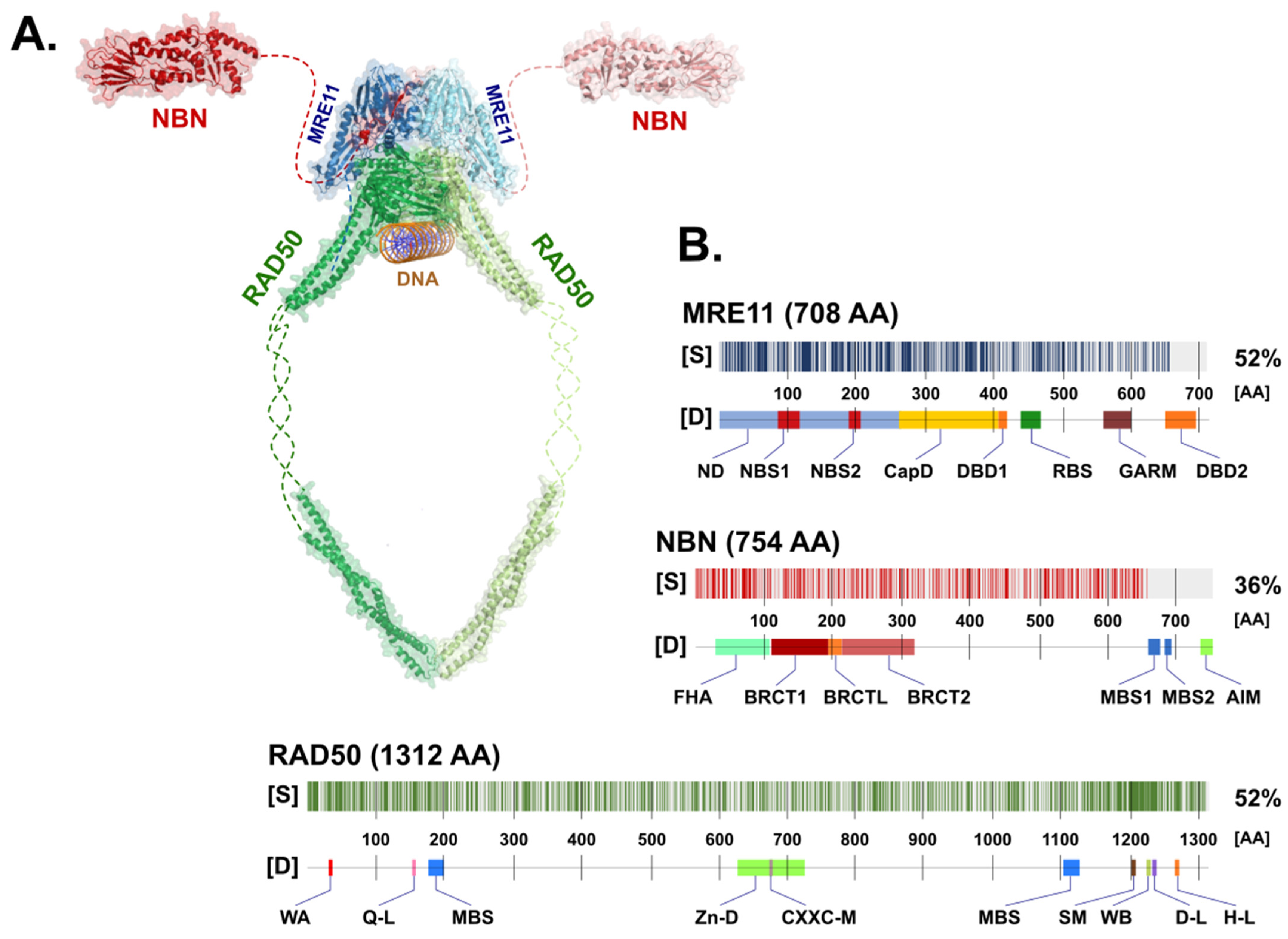
2. Structure of the MRN Complex
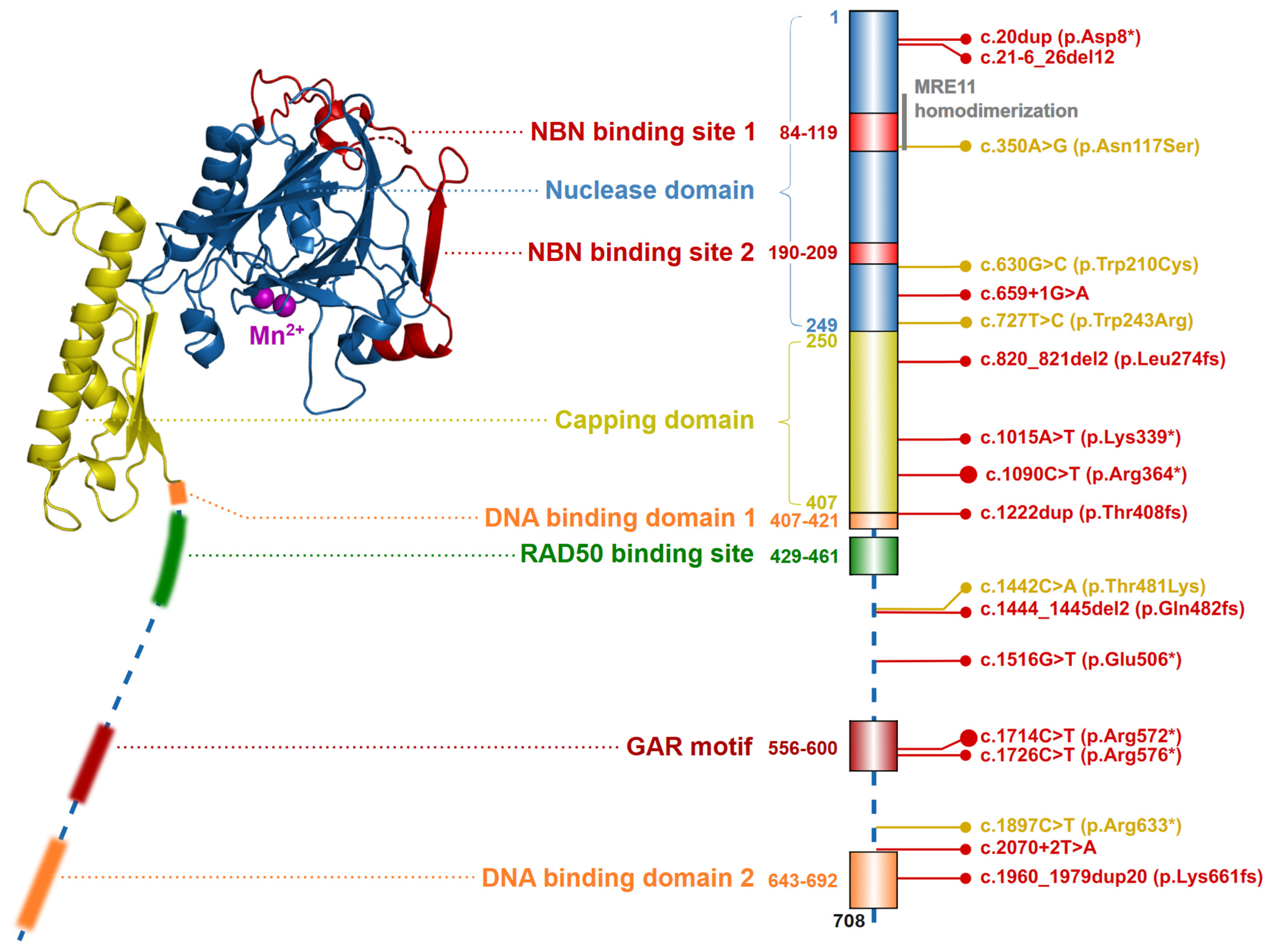
2.1. The Nuclease MRE11
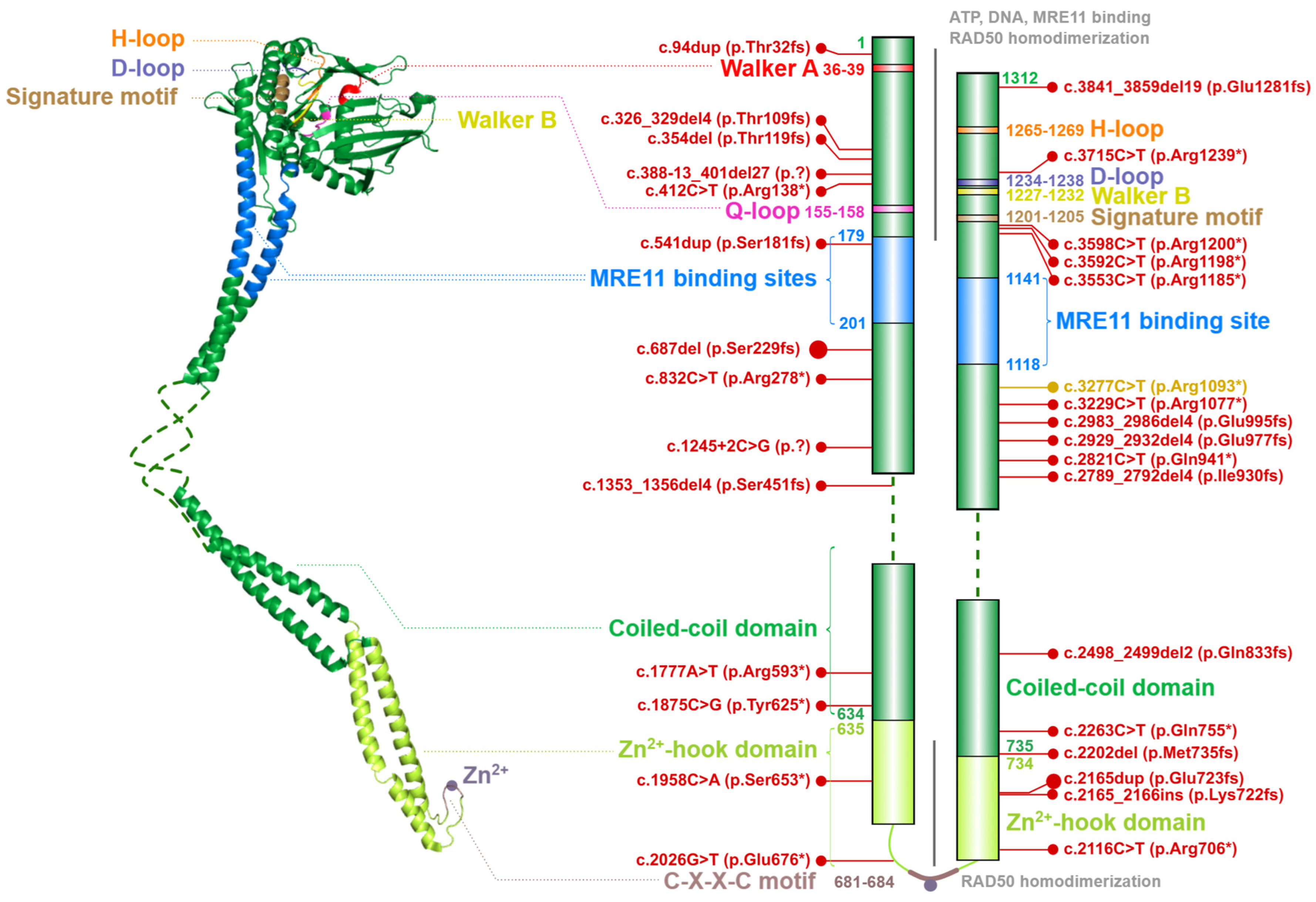
2.2. The RAD50 ATPase
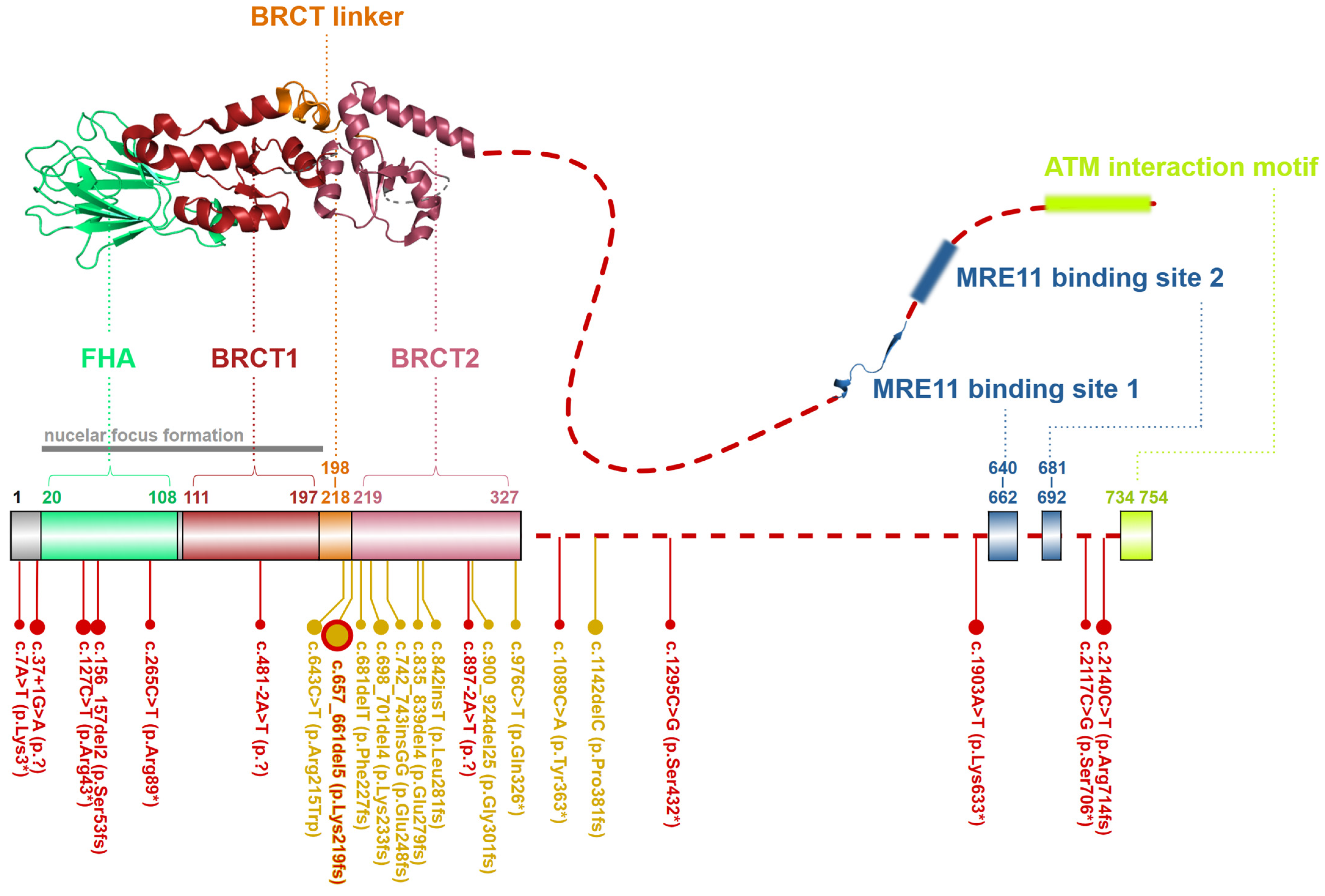
2.3. NBN, a Dynamic Connector
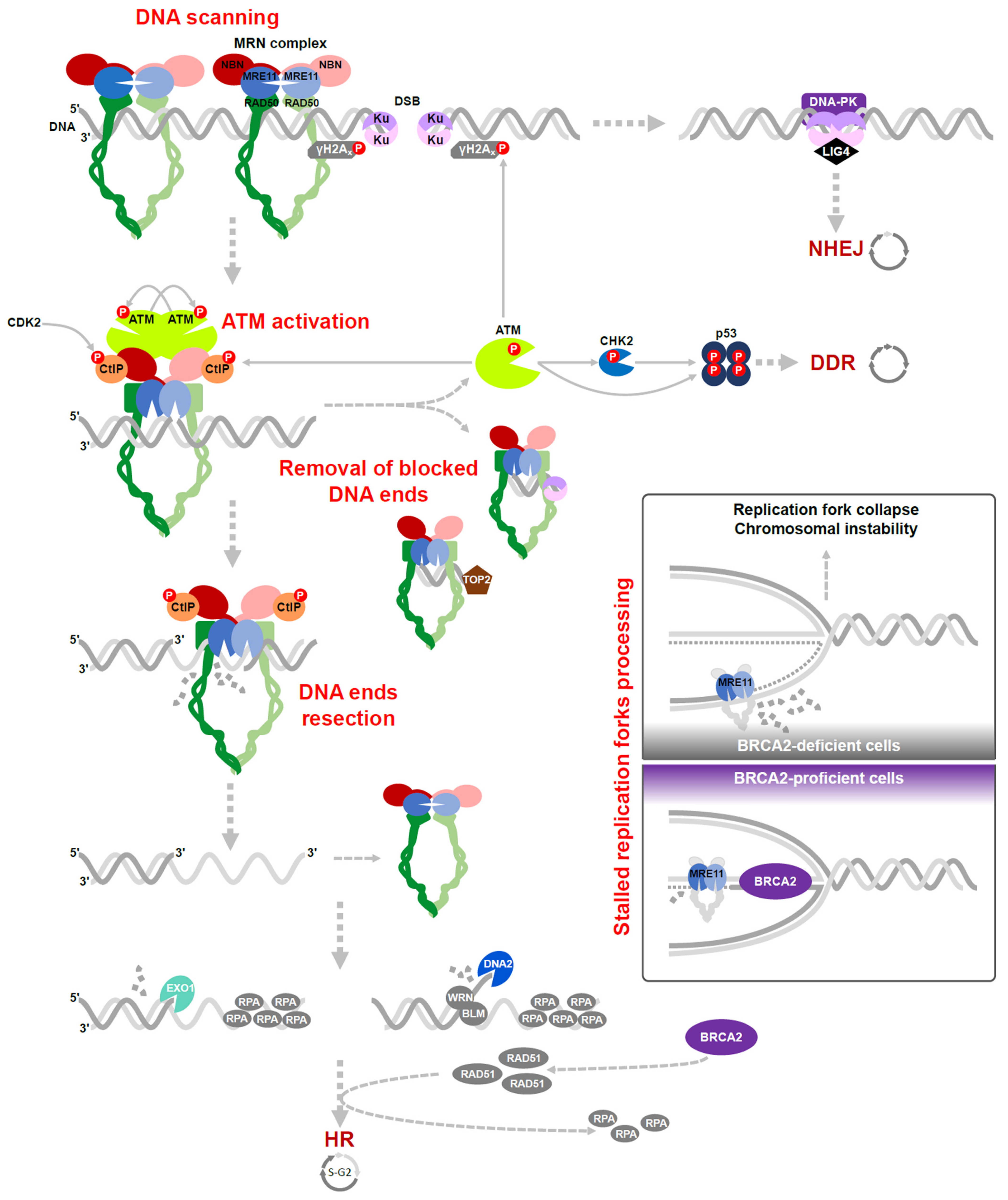
3. The MRN Complex Function in DSB Repair
3.1. ATM Activation
3.2. Removal of Blocked DNA Ends
3.3. Resection of DNA Ends
3.4. Processing of the Stalled Replication Forks
4. Germline Alterations of MRN Complex Genes in Autosomal Recessive Syndromes
4.1. The Nijmegen Breakage Syndrome (NBS)
4.2. Ataxia-Telangiectasia-like Disorder (ATLD)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nijmegen Breakage Syndrome (NBS) | NBS-like Disease (NBSLD) | Ataxia Telangiectasia-like Disease (ATLD) | ||
|---|---|---|---|---|
| Gene | NBN | RAD50 | MRE11 | |
| Inheritance | AR | AR | AR | |
| Described syndromic individuals | >1000 | 2 | ~30 | |
| Common features | Chromosomal instability | Yes | Yes | Yes |
| Ionizing radiation hypersensitivity | Yes | Yes | Yes | |
| Intellectual disability | Mild- moderate | Yes | Variable (limited evidence) | |
| Less common features | Microcephaly | Yes | Yes | No/Yes * |
| Short stature | Yes | Yes | No | |
| Craniofacial dysmorphism | Yes | Yes | No | |
| Unique features | Immunodeficiency | Yes | No | No |
| Increased rick (especially lymphoid tumors) | Yes | No | No | |
| Cerebellar ataxia/oculomotor apraxia | No | No | Yes | |
| Other features | Telangiectasia | No | No | No |
| AFP level | Normal | Normal | Normal | |
4.3. Nijmegen Breakage Syndrome-like Disorder (NBSLD)
5. Heterozygous Germline Alterations of MRN Complex Genes in Cancer Predisposition
5.1. Heterozygous Germline Variants in NBN
5.2. Heterozygous Germline Variants in MRE11 and RAD50
| Malignancy | Country | Patients (%) * | Controls (%) * | OR (95% CI); p-Value ** | Ref. |
|---|---|---|---|---|---|
| Brain | PL | 3/104 (2.9) | 74/12484 (0.6) | 4.9 (4.4–5.3); 0.003 | Ciara 2010 [96] |
| PL | 6/102 (5.9) | 0/300 (0) | 40.5 (2.3–721.2); <0.001 | Trubicka 2017 [105] | |
| Breast | PL | 5/230 (2.2) | 3/530 (0.6) | 3.9 (0.9–16.4); 0.06 | Gorski 2003 [106] |
| PL | 17/2012 (0.8) | 18/4000 (0.5) | 1.9 (1.0–3.7); 0.09 | Gorski 2006 [106] | |
| PL | 2/181 (1.1) | 21/4000 (0.5) | 2.1 (0.5–9.1); 0.6 | Kanka 2007 [107] | |
| PL | 4/224 (1.8) | 10/1620 (0.6) | 2.9 (0.9–9.4); 0.08 | Steffen 2004 [94] | |
| PL | 2/270 (0.7) | 2/295 (0.7) | 1.1 (0.2–7.9); 1.0 | Roznowski 2008 [108] | |
| US | 48/28,536 (0.2) | 39/26,264 (0.1) | 1.1 (0.7–1.8); 0.59 | Couch 2017 [109] | |
| DE | 12/5589 (0.2) | 9/2189 (0.4) | 0.5 (0.2–1.2); 0.15 | Hauke 2018 [110] | |
| PL | 18/2464 (0.7) | 22/4000 (0.6) | 1.3 (0.7–2.5); 0.46 | Rogoża-Janiszewska 2020 [111] | |
| US | 57/32,247 (0.2) | 51/32,544 (0.2) | 1.1 (0.7–1.6); 0.81 | Hu 2021 [91] | |
| CN | 6/8067 (0.07) | 5/13,129 (0.04) | 2.0 (0.6–6.4); 0.35 | Fu 2021 [112] | |
| US | 53/26,384 (0.20) | 115/64,649 (0.18) | 1.3 (0.9–1.8); 0.14 | Kurian 2017 [113] | |
| CZ | 8/703 (1.1) | 9/915 (1.0) | 1.2 (0.5–3.0); 0.81 | Mateju 2012 [114] | |
| Colorectum | PL | 3/234 (1.3) | 10/1620 (0.6) | 2.1 (0.6–7.7); 0.22 | Steffen 2004 [94] |
| CZ | 3/750 (0.4) | 5/1411 (0.35) | 0.95 (0.2–4.2); 0.95 | Pardini 2009 [115] | |
| Lymphoid | RU | 2/68 (2.9) | 0/548 (0) | 41.2 (1.9–862.9); 0.01 | Resnick 2003 [116] |
| PL | 2/42 (4.8) | 10/1620 (0.6) | 8.1 (1.7–37.9); 0.03 | Steffen 2004 [94] | |
| Melanoma | PL | 4/105 (3.8) | 10/1620 (0.6) | 6.4 (1.9–20.7); 0.008 | Steffen 2004 [94] |
| CZ | 7/264 (2.7) | 4/1479 (0.3) | 10.0 (2.5–47.0); <0.001 | Stolarova 2020 [117] | |
| Ovarian | US | 9/3257 (0.3) | 8/3447 (0.2) | 1.2 (0.5–3.1); 0.97 | Ramus 2015 [118] |
| CZ | 14/1320 (1.1) | 7/2278 (0.3) | 3.5 (1.3–10.2); 0.006 | Lhotova 2020 [119] | |
| US | 17/5020 (0.34) | 115/64,649 (0.18) | 1.85 (1.1–3.2); 0.03 | Kurian 2017 [113] | |
| Pancreas | PL | 8/383 (2.1) | 22/4000 (0.6) | 3.8 (1.7–8.6); 0.002 | Lener 2016 [120] |
| CZ | 5/241 (2.1) | 2/915 (0.2) | 9.7 (1.9–50.2); 0.006 | Borecka 2016 [121] | |
| Prostate | PL | 9/340 (2.6) | 9/1500 (0.6) | 4.5 (1.7–11.5); 0.002 | Cybulski 2004 [122] |
| US/FI/DE | 5/2127 (0.2) | 0/697 (0) | 3.61 (0.2–65.3); 0.58 | Hebbring 2006 [123] | |
| PL | 63/4162 (1.5) | 23/3956 (0.6) | 2.6 (1.6–4.3); <0.001 | Cybulski 2013 [124] | |
| PL | 11/390 (2.8) | 3/308 (0.9) | 3.0 (0.8–10.7); 0.1 | Wokołorczyk 2020 [125] |
6. Somatic Alterations in MRN Complex Genes in Tumors
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qiu, S.; Huang, J. MRN complex is an essential effector of DNA damage repair. J. Zhejiang Univ. B 2021, 22, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Macurek, L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libri, A.; Marton, T.; Deriano, L. The (Lack of) DNA Double-Strand Break Repair Pathway Choice during V(D)J Recombination. Front. Genet. 2022, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lingg, L.; Rottenberg, S.; Francica, P. Meiotic Genes and DNA Double Strand Break Repair in Cancer. Front. Genet. 2022, 13, 831620. [Google Scholar] [CrossRef]
- van den Bosch, M.; Bree, R.T.; Lowndes, N.F. The MRN complex: Coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003, 4, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.; Povirk, L.F. End-processing nucleases and phosphodiesterases: An elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair 2016, 43, 57–68. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell 2018, 71, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.; A Tainer, J. The MRE11–RAD50–NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Williams, R.S.; Williams, J.S.; Tainer, J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 2007, 85, 509–520. [Google Scholar] [CrossRef]
- Rupnik, A.; Grenon, M.; Lowndes, N. The MRN complex. Curr. Biol. 2008, 18, R455–R457. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Dunphy, W.G. The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Mol. Biol. Cell 2013, 24, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotheneder, M.; Stakyte, K.; van de Logt, E.; Bartho, J.D.; Lammens, K.; Fan, Y.; Alt, A.; Kessler, B.; Jung, C.; Roos, W.P.; et al. Cryo-EM structure of the Mre11-Rad50-Nbs1 complex reveals the molecular mechanism of scaffolding functions. Mol. Cell 2023, 83, 167–185.e9. [Google Scholar] [CrossRef] [PubMed]
- Schiller, C.B.; Lammens, K.; Guerini, I.; Coordes, B.; Feldmann, H.; Schlauderer, F.; Möckel, C.; Schele, A.; Strässer, K.; Jackson, S.P.; et al. Structure of Mre11–Nbs1 complex yields insights into ataxia-telangiectasia–like disease mutations and DNA damage signaling. Nat. Struct. Mol. Biol. 2012, 19, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Schiller, C.B.; Seifert, F.U.; Linke-Winnebeck, C.; Hopfner, K.-P. Structural Studies of DNA End Detection and Resection in Homologous Recombination. Cold Spring Harb. Perspect. Biol. 2014, 6, a017962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.M.; Iwasaki, H.; Russell, P.; et al. Nbs1 Flexibly Tethers Ctp1 and Mre11-Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.S.; Kim, J.S.; Park, Y.B.; Gwon, G.S.; Cho, Y. Crystal structure of the Mre11-Rad50-ATPγS complex: Understanding the interplay between Mre11 and Rad50. Genes Dev. 2011, 25, 1091–1104. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.C.; Padget, K.; Curtis, H.; Cowell, I.; Moiani, D.; Sondka, Z.; Morris, N.; Jackson, G.H.; Cockell, S.; Tainer, J.; et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 2012, 1, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Lafrance-Vanasse, J.; Williams, G.J.; Tainer, J.A. Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair. Prog. Biophys. Mol. Biol. 2015, 117, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Hopfner, K.-P.; Karcher, A.; Craig, L.; Woo, T.T.; Carney, J.P.; Tainer, J.A. Structural Biochemistry and Interaction Architecture of the DNA Double-Strand Break Repair Mre11 Nuclease and Rad50-ATPase. Cell 2001, 105, 473–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacho, E.J.; Maizels, N. DNA repair factor MRE11/RAD50 cleaves 3′-phosphotyrosyl bonds and resects DNA to repair damage caused by topoisomerase 1 poisons. J. Biol. Chem. 2011, 286, 44945–44951. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.B.; Chae, J.; Kim, Y.C.; Cho, Y. Crystal Structure of Human Mre11: Understanding Tumorigenic Mutations. Structure 2011, 19, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 Dimers Coordinate DNA End Bridging and Nuclease Processing in Double-Strand-Break Repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhang, H.; Jiang, Y.-N.; Wang, Z.-Q.; Sun, L.; Zhou, Z.-W. Post-Translational Modification of MRE11: Its Implication in DDR and Diseases. Genes 2021, 12, 1158. [Google Scholar] [CrossRef]
- Déry, U.; Coulombe, Y.; Rodrigue, A.; Stasiak, A.; Richard, S.; Masson, J.-Y. A Glycine-Arginine Domain in Control of the Human MRE11 DNA Repair Protein. Mol. Cell. Biol. 2008, 28, 3058–3069. [Google Scholar] [CrossRef] [Green Version]
- Gobbini, E.; Cassani, C.; Villa, M.; Bonetti, D.; Longhese, M.P. Functions and regulation of the MRX complex at DNA double-strand breaks. Microb. Cell 2016, 3, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabolotnaya, E.; Mela, I.; Henderson, R.M.; Robinson, N.P. Turning the Mre11/Rad50 DNA repair complex on its head: Lessons from SMC protein hinges, dynamic coiled-coil movements and DNA loop-extrusion? Biochem. Soc. Trans. 2020, 48, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Rojowska, A.; Lammens, K.; Seifert, F.U.; Direnberger, C.; Feldmann, H.; Hopfner, K. Structure of the Rad50 DNA double-strand break repair protein in complex with DNA. EMBO J. 2014, 33, 2847–2859. [Google Scholar] [CrossRef] [Green Version]
- Moncalian, G.; Lengsfeld, B.; Bhaskara, V.; Hopfner, K.-P.; Karcher, A.; Alden, E.; Tainer, J.A.; Paull, T.T. The Rad50 Signature Motif: Essential to ATP Binding and Biological Function. J. Mol. Biol. 2004, 335, 937–951. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Craig, L.; Moncalian, G.; Zinkel, R.A.; Usui, T.; Owen, B.A.L.; Karcher, A.; Henderson, B.; Bodmer, J.-L.; McMurray, C.T.; et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 2002, 418, 562–566. [Google Scholar] [CrossRef]
- Remali, J.; Aizat, W.M.; Ng, C.L.; Lim, Y.C.; Mohamed-Hussein, Z.-A.; Fazry, S. In silico analysis on the functional and structural impact of Rad50 mutations involved in DNA strand break repair. PeerJ 2020, 8, e9197. [Google Scholar] [CrossRef]
- Moreno-Herrero, F.; de Jager, M.; Dekker, N.H.; Kanaar, R.; Wyman, C.; Dekker, C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature 2005, 437, 440–443. [Google Scholar] [CrossRef]
- Park, Y.B.; Hohl, M.; Padjasek, M.; Jeong, E.; Jin, K.S.; Krezel, A.; Petrini, M.H.J.H.J.; Cho, Y.B.P.E.J.Y. Eukaryotic Rad50 functions as a rod-shaped dimer. Nat. Struct. Mol. Biol. 2017, 24, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, F.U.; Lammens, K.; Stoehr, G.; Kessler, B.; Hopfner, K. Structural mechanism of ATP-dependent DNA binding and DNA end bridging by eukaryotic Rad50. EMBO J. 2016, 35, 759–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Käshammer, L.; Saathoff, J.-H.; Lammens, K.; Gut, F.; Bartho, J.; Alt, A.; Kessler, B.; Hopfner, K.-P. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell 2019, 76, 382–394.e6. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanová, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a Novel DNA Double-Strand Break Repair Protein, Is Mutated in Nijmegen Breakage Syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilli, D.; Mirasole, C.; Pennisi, R.; Pallotta, V.; D′Alessandro, A.; Antoccia, A.; Zolla, L.; Ascenzi, P.; di Masi, A. Identification of the Interactors of Human Nibrin (NBN) and of Its 26 kDa and 70 kDa Fragments Arising from the NBN 657del5 Founder Mutation. PLoS ONE 2014, 9, e114651. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.R.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Ohara, M.; Seki, R.; Tauchi, H. Dancing on damaged chromatin: Functions of ATM and the RAD50/MRE11/NBS1 complex in cellular responses to DNA damage. J. Radiat. Res. 2008, 49, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Morgen, P.; Burdova, K.; Flower, T.G.; O′Reilly, N.J.; Boulton, S.J.; Smerdon, S.J.; Macurek, L.; Hořejší, Z. MRE11 stability is regulated by CK2-dependent interaction with R2TP complex. Oncogene 2017, 36, 4943–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R., 3rd; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Grosbart, M.; Anand, R.; Wyman, C.; Cejka, P.; Petrini, J.H. The Mre11-Nbs1 Interface Is Essential for Viability and Tumor Suppression. Cell Rep. 2017, 18, 496–507. [Google Scholar] [CrossRef]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhou, Z.; Yang, H.; Wang, W. MRE11-RAD50-NBS1-CtIP: One key nuclease ensemble functions in the maintenance of genome stability. Genome Instab. Dis. 2022, 3, 123–135. [Google Scholar] [CrossRef]
- Wojtaszek, J.L.; Williams, R.S. The ends in sight: Mre11-Rad50-Nbs1 complex structures come into focus. Mol. Cell 2023, 83, 160–162. [Google Scholar] [CrossRef]
- Sevcik, J.; Falk, M.; Kleiblova, P.; Lhota, F.; Stefancikova, L.; Janatova, M.; Weiterova, L.; Lukasova, E.; Kozubek, S.; Pohlreich, P.; et al. The BRCA1 alternative splicing variant Δ14-15 with an in-frame deletion of part of the regulatory serine-containing domain (SCD) impairs the DNA repair capacity in MCF-7 cells. Cell. Signal. 2012, 24, 1023–1030. [Google Scholar] [CrossRef]
- Prokopcova, J.; Kleibl, Z.; Banwell, C.M.; Pohlreich, P. The role of ATM in breast cancer development. Breast Cancer Res. Treat. 2006, 104, 121–128. [Google Scholar] [CrossRef]
- Ueno, S.; Sudo, T.; Hirasawa, A. ATM: Functions of ATM Kinase and Its Relevance to Hereditary Tumors. Int. J. Mol. Sci. 2022, 23, 523. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.R. The DNA Double-Strand Break Repair Gene hMRE11 Is Mutated in Individuals with an Ataxia-Telangiectasia-like Disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [Green Version]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.-Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 2016, 64, 580–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buis, J.; Stoneham, T.; Spehalski, E.; Ferguson, D.O. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol. 2012, 19, 246–252. [Google Scholar] [CrossRef]
- Paull, T.T.; Gellert, M. The 3′ to 5′ Exonuclease Activity of Mre11 Facilitates Repair of DNA Double-Strand Breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Oh, J.-M.; Myung, K. Crosstalk between different DNA repair pathways for DNA double strand break repairs. Mutat. Res. Toxicol. Environ. Mutagen. 2021, 873, 503438. [Google Scholar] [CrossRef] [PubMed]
- Elkholi, I.E.; Foulkes, W.D.; Rivera, B. MRN Complex and Cancer Risk: Old Bottles, New Wine. Clin. Cancer Res. 2021, 27, 5465–5471. [Google Scholar] [CrossRef]
- Roy, S.; Tomaszowski, K.-H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLθ pathways. Elife 2018, 7, e31723. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Borsos, B.N.; Majoros, H.; Pankotai, T. Ubiquitylation-Mediated Fine-Tuning of DNA Double-Strand Break Repair. Cancers 2020, 12, 1617. [Google Scholar] [CrossRef]
- Reginato, G.; Cejka, P. The MRE11 complex: A versatile toolkit for the repair of broken DNA. DNA Repair 2020, 91–92, 102869. [Google Scholar] [CrossRef]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Prim. 2019, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.S.; Zinkel, R.; Petrini, J.H. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat. Genet. 2001, 27, 417–421. [Google Scholar] [CrossRef]
- Varon, R.; Dutrannoy, V.; Weikert, G.; Tanzarella, C.; Antoccia, A.; Stöckl, L.; Spadoni, E.; Krüger, L.-A.; di Masi, A.; Sperling, K.; et al. Mild Nijmegen breakage syndrome phenotype due to alternative splicing. Hum. Mol. Genet. 2006, 15, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Bagińska, B.; Kalina, M.A.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Seemanova, E.; Varon, R.; Vejvalka, J.; Jarolim, P.; Seeman, P.; Chrzanowska, K.H.; Digweed, M.; Resnick, I.; Kremensky, I.; Saar, K.; et al. The Slavic NBN Founder Mutation: A Role for Reproductive Fitness? PLoS ONE 2016, 11, e0167984. [Google Scholar] [CrossRef] [Green Version]
- Salewsky, B.; Hildebrand, G.; Rothe, S.; Parplys, A.C.; Radszewski, J.; Kieslich, M.; Wessendorf, P.; Krenzlin, H.; Borgmann, K.; Nussenzweig, A.; et al. Directed Alternative Splicing in Nijmegen Breakage Syndrome: Proof of Principle Concerning Its Therapeutical Application. Mol. Ther. 2016, 24, 117–124. [Google Scholar] [CrossRef]
- Dumon-Jones, V.; Frappart, P.-O.; Tong, W.-M.; Sajithlal, G.; Hulla, W.; Schmid, G.; Herceg, Z.; Digweed, M.; Wang, Z.-Q. Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res. 2003, 63, 7263–7269. [Google Scholar]
- Desjardins, S.; Beauparlant, J.C.; Labrie, Y.; Ouellette, G.; INHERIT BRCAs; Francine Durocher. Variations in the NBN/NBS1 gene and the risk of breast cancer in non-BRCA1/2 French Canadian families with high risk of breast cancer. BMC Cancer 2009, 9, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varon, R.; Seemanova, E.; Chrzanowska, K.; Hnateyko, O.; Piekutowska-Abramczuk, D.; Krajewska-Walasek, M.; Sykut-Cegielska, J.; Sperling, K.; Reis, A. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. Eur. J. Hum. Genet. 2000, 8, 900–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolska-Kusnierz, B.; Pastorczak, A.; Fendler, W.; Wakulinska, A.; Dembowska-Baginska, B.; Heropolitanska-Pliszka, E.; Piątosa, B.; Pietrucha, B.; Kałwak, K.; Ussowicz, M.; et al. Hematopoietic Stem Cell Transplantation Positively Affects the Natural History of Cancer in Nijmegen Breakage Syndrome. Clin. Cancer Res. 2021, 27, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Dembowska-Baginska, B.; Perek, D.; Brozyna, A.; Wakulinska, A.; Olczak-Kowalczyk, D.; Gladkowska-Dura, M.; Grajkowska, W.; Chrzanowska, K.H. Non-Hodgkin lymphoma (NHL) in children with Nijmegen Breakage syndrome (NBS). Pediatr. Blood Cancer 2008, 52, 186–190. [Google Scholar] [CrossRef]
- Pollard, J.M.; Gatti, R.A. Clinical Radiation Sensitivity with DNA Repair Disorders: An Overview. Int. J. Radiat. Oncol. 2009, 74, 1323–1331. [Google Scholar] [CrossRef] [Green Version]
- Hasbaoui, B.E.; Elyajouri, A.; Abilkassem, R.; Agadr, A. Nijmegen breakage syndrome: Case report and review of literature. Pan. Afr. Med. J. 2020, 35, 85. [Google Scholar] [CrossRef]
- Pasic, S.; Vujic, D.; Fiorini, M.; Notarangelo, L.D. T-cell lymphoblastic leukemia/lymphoma in Nijmegen breakage syndrome. Haematologica 2004, 89, ECR27. [Google Scholar]
- Rahman, S.; Canny, M.D.; Buschmann, T.A.; Latham, M.P. A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex. Cells 2020, 9, 1678. [Google Scholar] [CrossRef]
- Seemanová, E.; Jarolim, P.; Seeman, P.; Varon, R.; Digweed, M.; Swift, M.; Sperling, K. Cancer Risk of Heterozygotes With the NBN Founder Mutation. Gynecol. Oncol. 2007, 99, 1875–1880. [Google Scholar] [CrossRef]
- Fiévet, A.; Bellanger, D.; Valence, S.; Mobuchon, L.; Afenjar, A.; Giuliano, F.; d′Enghien, C.D.; Parfait, B.; Pedespan, J.M.; Auger, N. Three new cases of ataxia-telangiectasia-like disorder: No impairment of the ATM pathway, but S-phase checkpoint defect. Hum. Mutat. 2019, 40, 1690–1699. [Google Scholar] [CrossRef]
- Ragamin, A.; Yigit, G.; Bousset, K.; Beleggia, F.; Verheijen, F.W.; de Wit, M.Y.; Strom, T.M.; Dörk, T.; Wollnik, B.; Mancini, G.M.S. Human RAD50 deficiency: Confirmation of a distinctive phenotype. Am. J. Med. Genet. Part A 2020, 182, 1378–1386. [Google Scholar] [CrossRef] [Green Version]
- Gueven, N.; Chen, P.; Nakamura, J.; Becherel, O.; Kijas, A.; Grattan-Smith, P.; Lavin, M. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience 2007, 145, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Mahale, R.R.; Reddy, N.; Mathuranth, P.; Mailankody, P.; Padmanabha, H.; Retnaswami, C.S. A rare case of ataxia-telangiectasia-like disorder with MRE11 mutation. J. Pediatr. Neurosci. 2020, 15, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Alsbeih, G. MRE11A Gene Mutations Responsible for the Rare Ataxia Telangiectasia-Like Disorder. In Human Genetic Diseases; IntechOpen: London, UK, 2011. [Google Scholar] [CrossRef] [Green Version]
- Shull, E.R.; Lee, Y.; Nakane, H.; Stracker, T.H.; Zhao, J.; Russell, H.R.; Petrini, J.H.; McKinnon, P.J. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev. 2009, 23, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Raslan, I.R.; Matos, P.C.A.P.; Ciarlariello, V.B.; Daghastanli, K.H.; Rosa, A.B.R.; Arita, J.H.; Aranda, C.S.; Barsottini, O.G.P.; Pedroso, J.L. Beyond Typical Ataxia Telangiectasia: How to Identify the Ataxia Telangiectasia-Like Disorders. Mov. Disord. Clin. Pract. 2020, 8, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Miyamoto, T.; Sakamoto, H.; Izumi, H.; Nakazawa, Y.; Ogi, T.; Tahara, H.; Oku, S.; Hiramoto, A.; Shiiki, T.; et al. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair 2011, 10, 314–321. [Google Scholar] [CrossRef]
- Uchisaka, N.; Takahashi, N.; Sato, M.; Kikuchi, A.; Mochizuki, S.; Imai, K.; Nonoyama, S.; Ohara, O.; Watanabe, F.; Mizutani, S.; et al. Two Brothers with Ataxia-Telangiectasia-like Disorder with Lung Adenocarcinoma. J. Pediatr. 2009, 155, 435–438. [Google Scholar] [CrossRef]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 Deficiency in a Nijmegen Breakage Syndrome-like Disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Breast Cancer Association, C.; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Breast Cancer Risk Genes-Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Shi, Z.; Lu, L.; Bs, W.K.R.; Yang, W.; Wei, J.; Wang, Q.; Engelmann, V.; Zheng, S.L.; Cooney, K.A.; Isaacs, W.B.; et al. Association of germline rare pathogenic mutations in guideline-recommended genes with prostate cancer progression: A meta-analysis. Prostate 2021, 82, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Varon, R.; Mosor, M.; Maneva, G.; Maurer, M.; Stumm, M.; Nowakowska, D.; Rubach, M.; Kosakowska, E.; Ruka, W.; et al. Increased cancer risk of heterozygotes withNBS1 germline mutations in poland. Int. J. Cancer 2004, 111, 67–71. [Google Scholar] [CrossRef]
- Horackova, K.F.; Zemankova, S.; Nehasil, P.; Cerna, P.; Neroldova, M.; Otahalova, M.; Kral, B.; Hovhannisyan, J.; Stranecky, M.; Zima, V.; et al. Low Frequency of Cancer-Predisposition Gene Mutations in Liver Transplant Candidates with Hepatocellular Carcinoma. Cancers 2022, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Ciara, E.; Piekutowska-Abramczuk, D.; Popowska, E.; Grajkowska, W.; Barszcz, S.; Perek, D.; Dembowska-Bagińska, B.; Perek-Polnik, M.; Kowalewska, E.; Czajńska, A.; et al. Heterozygous germ-line mutations in the NBN gene predispose to medulloblastoma in pediatric patients. Acta Neuropathol. 2009, 119, 325–334. [Google Scholar] [CrossRef]
- Soucek, P.; Gut, I.; Trneny, M.; Skovlund, E.; Alnaes, G.G.; Kristensen, T.; Børresen-Dale, A.-L.; Kristensen, V.N. Multiplex single-tube screening for mutations in the Nijmegen Breakage Syndrome (NBS1) gene in Hodgkin′s and non-Hodgkin′s lymphoma patients of Slavic origin. Eur. J. Hum. Genet. 2003, 11, 416–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belhadj, S.; Terradas, M.; Munoz-Torres, P.M.; Aiza, G.; Navarro, M.; Capellá, G.; Valle, L. Candidate genes for hereditary colorectal cancer: Mutational screening and systematic review. Hum. Mutat. 2020, 41, 1563–1576. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Iijima, T.; Mori, T.; Takahashi, K.; Matsumoto, H.; Miyamoto, H.; Hishima, T.; Miyaki, M. Accumulation Profile of Frameshift Mutations during Development and Progression of Colorectal Cancer from Patients with Hereditary Nonpolyposis Colorectal Cancer. Dis. Colon Rectum 2006, 49, 399–406. [Google Scholar] [CrossRef]
- Wang, X.; Szabo, C.; Qian, C.; Amadio, P.G.; Thibodeau, S.N.; Cerhan, J.R.; Petersen, G.M.; Liu, W.; Couch, F.J. Mutational Analysis of Thirty-two Double-Strand DNA Break Repair Genes in Breast and Pancreatic Cancers. Cancer Res. 2008, 68, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Heikkinen, K.; Rapakko, K.; Karppinen, S.-M.; Erkko, H.; Knuutila, S.; Lundán, T.; Mannermaa, A.; Børresen-Dale, A.-L.; Borg, Å.; Barkardottir, R.B.; et al. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis 2005, 27, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Rostami, P.; Zendehdel, K.; Shirkoohi, R.; Ebrahimi, E.; Ataei, M.; Imanian, H.; Najmabadi, H.; Akbari, M.R.; Sanati, M.H. Gene Panel Testing in Hereditary Breast Cancer. Arch. Iran. Med. 2020, 23, 155–162. [Google Scholar] [PubMed]
- Damiola, F.; Pertesi, M.; Oliver, J.; Calvez-Kelm, F.L.; Voegele, C.; Young, E.L.; Robinot, N.; Forey, N.; Durand, G.; Vallée, M.P. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: Results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 2014, 16, R58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhrhammer, N.; Delort, L.; Bignon, Y.-J. Rad50 c.687delT Does Not Contribute Significantly to Familial Breast Cancer in a French Population. Cancer Epidemiol. Biomark. Prev. 2009, 18, 684–685. [Google Scholar] [CrossRef] [Green Version]
- Trubicka, J.; Żemojtel, T.; Hecht, J.; Falana, K.; Abramczuk, D.P.; Płoski, R.; Perek-Polnik, M.; Drogosiewicz, M.; Grajkowska, W.; Ciara, E.; et al. The germline variants in DNA repair genes in pediatric medulloblastoma: A challenge for current therapeutic strategies. BMC Cancer 2017, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Górski, B.; Dębniak, T.; Masojć, B.; Mierzejewski, M.; Mędrek, K.; Cybulski, C.; Jakubowska, A.; Kurzawski, G.; Chosia, M.; Scott, R.; et al. Germline 657del5 mutation in the NBS1 gene in breast cancer patients. Int. J. Cancer 2003, 106, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Kanka, C.; Brozek, I.; Skalska, B.; Siemiatkowska, A.; Limon, J. Germline NBS1 mutations in families with aggregation of Breast and/or ovarian cancer from north-east Poland. Anticancer. Res. 2007, 27, 3015–3018. [Google Scholar]
- Rożnowski, K.; Januszkiewicz-Lewandowska, D.; Mosor, M.; Pernak, M.; Litwiniuk, M.; Nowak, J. I171V germline mutation in the NBS1 gene significantly increases risk of breast cancer. Breast Cancer Res. Treat. 2007, 110, 343–348. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef]
- Rogoża-Janiszewska, E.; Malińska, K.; Cybulski, C.; Jakubowska, A.; Gronwald, J.; Huzarski, T.; Lener, M.; Górski, B.; Kluźniak, W.; Rudnicka, H.; et al. Prevalence of Recurrent Mutations Predisposing to Breast Cancer in Early-Onset Breast Cancer Patients from Poland. Cancers 2020, 12, 2321. [Google Scholar] [CrossRef]
- Fu, F.; Zhang, D.; Hu, L.; Sundaram, S.; Ying, D.; Zhang, Y.; Fu, S.; Zhang, J.; Yao, L.; Xu, Y. Association between 15 known or potential breast cancer susceptibility genes and breast cancer risks in Chinese women. Cancer Biol. Med. 2021, 19, 253–262. [Google Scholar] [CrossRef]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.-R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived from Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Mateju, M.; Kleiblova, P.; Kleibl, Z.; Janatova, M.; Soukupova, J.; Tichá, I.; Novotny, J.; Pohlreich, P. Germline mutations 657del5 and 643C>T (R215W) in NBN are not likely to be associated with increased risk of breast cancer in Czech women. Breast Cancer Res. Treat. 2012, 133, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Naccarati, A.; Polakova, V.; Smerhovsky, Z.; Hlavata, I.; Soucek, P.; Novotny, J.; Vodickova, L.; Tomanova, V.; Landi, S.; et al. NBN 657del5 heterozygous mutations and colorectal cancer risk in the Czech Republic. Mutat. Res. Mol. Mech. Mutagen. 2009, 666, 64–67. [Google Scholar] [CrossRef]
- Resnick, I.B.; Kondratenko, I.; Pashanov, E.; Maschan, A.A.; Karachunsky, A.; Togoev, O.; Timakov, A.; Polyakov, A.; Tverskaya, S.; Evgrafov, O.; et al. 657del5 mutation in the gene for Nijmegen breakage syndrome (NBS1) in a cohort of Russian children with lymphoid tissue malignancies and controls. Am. J. Med. Genet. 2003, 120A, 174–179. [Google Scholar] [CrossRef]
- Stolarova, L.; Jelinkova, S.; Storchova, R.; Machackova, E.; Zemankova, P.; Vocka, M.; Kodet, O.; Kral, J.; Cerna, M.; Volkova, Z.; et al. Identification of Germline Mutations in Melanoma Patients with Early Onset, Double Primary Tumors, or Family Cancer History by NGS Analysis of 217 Genes. Biomedicines 2020, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhotova, K.; Stolarova, L.; Zemankova, P.; Vocka, M.; Janatova, M.; Borecka, M.; Cerna, M.; Jelinkova, S.; Kral, J.; Volkova, Z.; et al. Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers 2020, 12, 956. [Google Scholar] [CrossRef]
- Lener, M.R.; Scott, R.J.; Kluźniak, W.; Baszuk, P.; Cybulski, C.; Wiechowska-Kozłowska, A.; Huzarski, T.; Byrski, T.; Kładny, J.; Pietrzak, S. Do founder mutations characteristic of some cancer sites also predispose to pancreatic cancer? Int. J. Cancer 2016, 139, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Borecka, M.; Zemankova, P.; Lhota, F.; Soukupova, J.; Kleiblova, P.; Vocka, M.; Soucek, P.; Ticha, I.; Kleibl, Z.; Janatova, M. The c.657del5 variant in the NBN gene predisposes to pancreatic cancer. Gene 2016, 587, 169–172. [Google Scholar] [CrossRef]
- Cybulski, C.; Górski, B.; Debniak, T.; Gliniewicz, B.; Mierzejewski, M.; Masojć, B.; Jakubowska, A.; Matyjasik, J.; Złowocka, E.; Sikorski, A.; et al. NBS1 Is a Prostate Cancer Susceptibility Gene. Cancer Res. 2004, 64, 1215–1219. [Google Scholar] [CrossRef] [Green Version]
- Hebbring, S.J.; Fredriksson, H.; White, K.A.; Maier, C.; Ewing, C.; McDonnell, S.K.; Jacobsen, S.J.; Cerhan, J.; Schaid, D.J.; Ikonen, T.; et al. Role of the Nijmegen Breakage Syndrome 1 Gene in Familial and Sporadic Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, C.; Wokołorczyk, D.; Kluźniak, W.; Jakubowska, A.; Górski, B.; Gronwald, J.; Huzarski, T.; Kashyap, A.; Byrski, T.; Dȩbniak, T.; et al. An inherited NBN mutation is associated with poor prognosis prostate cancer. Br. J. Cancer 2013, 108, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wokołorczyk, D.; Kluźniak, W.; Huzarski, T.; Gronwald, J.; Szymiczek, A.; Rusak, B.; Stempa, K.; Gliniewicz, K.; Kashyap, A.; Morawska, S. Mutations in ATM, NBN and BRCA2 predispose to aggressive prostate cancer in Poland. Int. J. Cancer 2020, 147, 2793–2800. [Google Scholar] [CrossRef]
- Panou, V.; Gadiraju, M.; Wolin, A.; Weipert, C.M.; Skarda, E.; Husain, A.N.; Patel, J.D.; Rose, B.; Zhang, S.R.; Weatherly, M.; et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J. Clin. Oncol. 2018, 36, 2863–2871. [Google Scholar] [CrossRef]
- Bartkova, J.; Tommiska, J.; Oplustilova, L.; Aaltonen, K.; Tamminen, A.; Heikkinen, T.; Mistrik, M.; Aittomäki, K.; Blomqvist, C.; Heikkilä, P.; et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol. Oncol. 2008, 2, 296–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.M.; Wang, H.-C.; Chen, S.-T.; Hsu, G.-C.; Shen, C.-Y.; Yu, J.-C. Breast cancer risk is associated with the genes encoding the DNA double-strand break repair Mre11/Rad50/Nbs1 complex. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2024–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castera, L.; Krieger, S.; Rousselin, A.; Legros, A.; Baumann, J.-J.; Bruet, O.; Brault, B.; Fouillet, R.; Goardon, N.; Letac, O.; et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur. J. Hum. Genet. 2014, 22, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkholi, I.E.; Di Iorio, M.; Fahiminiya, S.; Arcand, S.L.; Han, H.; Nogué, C.; Behl, S.; Hamel, N.; Giroux, S.; de Ladurantaye, M.; et al. Investigating the causal role of MRE11A p.E506* in breast and ovarian cancer. Sci. Rep. 2021, 11, 2409. [Google Scholar] [CrossRef]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Bs, S.G.; Hart, S.N.; Yadav, S.; Hu, C.; Na, J.; Goldgar, D.E.; et al. A clinical guide to hereditary cancer panel testing: Evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Anesth. Analg. 2019, 22, 407–415. [Google Scholar] [CrossRef] [Green Version]
- McGuigan, A.; Whitworth, J.; Andreou, A.; Hearn, T.; Ambrose, J.C.; Arumugam, P.; Bevers, R.; Bleda, M.; Boardman-Pretty, F.; Boustred, C.R.; et al. Multilocus Inherited Neoplasia Allele Syndrome (MINAS): An update. Eur. J. Hum. Genet. 2022, 30, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, S.; Chang, P. New PARP targets for cancer therapy. Nat. Rev. Cancer 2014, 14, 502–509. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, G.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.-D.; Limagne, E.; Thibaudin, M.; Truntzer, C.; Bertaut, A.; Rederstorff, E.; Ghiringhelli, F. Precision medicine phase II study evaluating the efficacy of a double immunotherapy by durvalumab and tremelimumab combined with olaparib in patients with solid cancers and carriers of homologous recombination repair genes mutation in response or stable after olaparib treatment. BMC Cancer 2020, 20, 748. [Google Scholar] [CrossRef]
- McPherson, M.T.; Holub, A.S.; Husbands, A.Y.; Petreaca, R.C. Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells. Cancers 2020, 12, 3794. [Google Scholar] [CrossRef]
- Al-Ahmadie, H.; Iyer, G.; Hohl, M.; Asthana, S.; Inagaki, A.; Schultz, N.; Hanrahan, A.J.; Scott, S.N.; Brannon, A.R.; McDermott, G.C.; et al. Synthetic Lethality in ATM-Deficient RAD50-Mutant Tumors Underlies Outlier Response to Cancer Therapy. Cancer Discov. 2014, 4, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Boswell, Z.K.; Canny, M.D.; Buschmann, T.A.; Sang, J.; Latham, M.P. Adjacent mutations in the archaeal Rad50 ABC ATPase D-loop disrupt allosteric regulation of ATP hydrolysis through different mechanisms. Nucleic Acids Res. 2019, 48, 2457–2472. [Google Scholar] [CrossRef] [Green Version]
- Seborova, K.; Hlavac, V.; Holy, P.; Bjørklund, S.S.; Fleischer, T.; Rob, L.; Hruda, M.; Bouda, J.; Mrhalova, M.; Allah, M.M.K.A.O.; et al. Complex molecular profile of DNA repair genes in epithelial ovarian carcinoma patients with different sensitivity to platinum-based therapy. Front. Oncol. 2022, 12, 1016958. [Google Scholar] [CrossRef]
- Chae, Y.K.; Anker, J.F.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Kalyan, A.; Santa-Maria, C.A.; Platanias, L.C.; Giles, F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget 2016, 7, 23312–23321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Li, S.; Tang, X.; Wang, Y.; Guo, W.; Cao, G.; Chen, K.; Zhang, M.; Guan, M.; Yang, D. Copy Number Amplification of DNA Damage Repair Pathways Potentiates Therapeutic Resistance in Cancer. Theranostics 2020, 10, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Berlin, A.; LaLonde, E.; Zafarana, G.; Sykes, J.; Lam, W.; Meng, A.; Milosevic, M.; Van der Kwast, T.; Boutros, P.; Bristow, R. PD-0300: NBN gain is predictive for adverse outcome following image-guided radiotherapy for localized prostate cancer. Radiother. Oncol. 2014, 111, S116–S117. [Google Scholar] [CrossRef]
- Loh, P.-R.; Genovese, G.; McCarroll, S.A. Monogenic and polygenic inheritance become instruments for clonal selection. Nature 2020, 584, 136–141. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otahalova, B.; Volkova, Z.; Soukupova, J.; Kleiblova, P.; Janatova, M.; Vocka, M.; Macurek, L.; Kleibl, Z. Importance of Germline and Somatic Alterations in Human MRE11, RAD50, and NBN Genes Coding for MRN Complex. Int. J. Mol. Sci. 2023, 24, 5612. https://doi.org/10.3390/ijms24065612
Otahalova B, Volkova Z, Soukupova J, Kleiblova P, Janatova M, Vocka M, Macurek L, Kleibl Z. Importance of Germline and Somatic Alterations in Human MRE11, RAD50, and NBN Genes Coding for MRN Complex. International Journal of Molecular Sciences. 2023; 24(6):5612. https://doi.org/10.3390/ijms24065612
Chicago/Turabian StyleOtahalova, Barbora, Zuzana Volkova, Jana Soukupova, Petra Kleiblova, Marketa Janatova, Michal Vocka, Libor Macurek, and Zdenek Kleibl. 2023. "Importance of Germline and Somatic Alterations in Human MRE11, RAD50, and NBN Genes Coding for MRN Complex" International Journal of Molecular Sciences 24, no. 6: 5612. https://doi.org/10.3390/ijms24065612
APA StyleOtahalova, B., Volkova, Z., Soukupova, J., Kleiblova, P., Janatova, M., Vocka, M., Macurek, L., & Kleibl, Z. (2023). Importance of Germline and Somatic Alterations in Human MRE11, RAD50, and NBN Genes Coding for MRN Complex. International Journal of Molecular Sciences, 24(6), 5612. https://doi.org/10.3390/ijms24065612