
Combination Therapies Targeting Apoptosis in Paediatric AML: Understanding the Molecular Mechanisms of AML Treatments Using Phosphoproteomics

 and
and 
Abstract
:1. Introduction
2. Results and Discussion
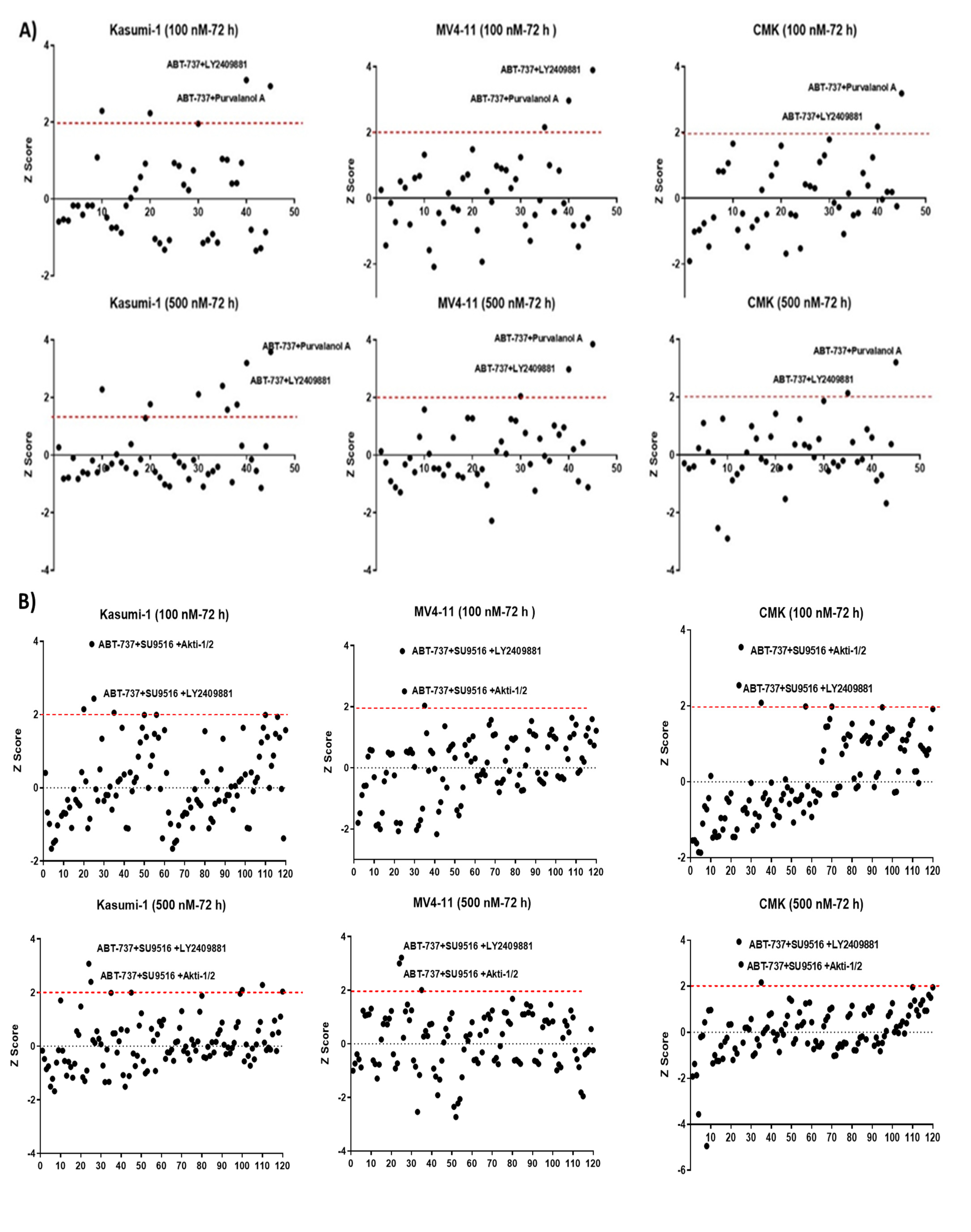
2.1. Deconvolution of 10 Drugs Identified One Consistent Double and Triple Combination across Multiple Paediatric Cell Lines as Potential Therapies for Paediatric AML
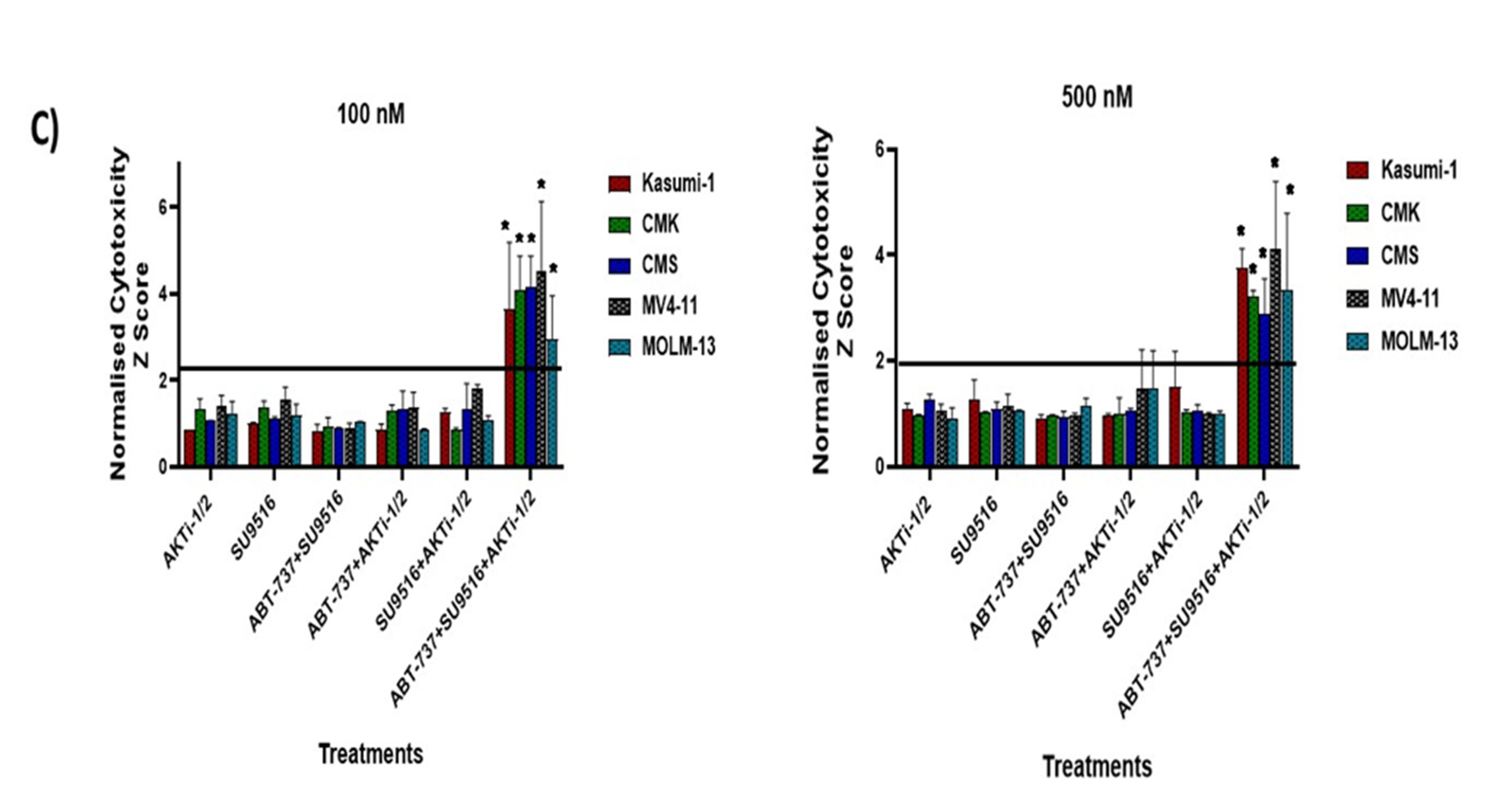
2.2. Validation of Double and Triple Combinations: ABT-737 + Purvalanol-A and ABT-737 + AKTi-1/2 + SU9516
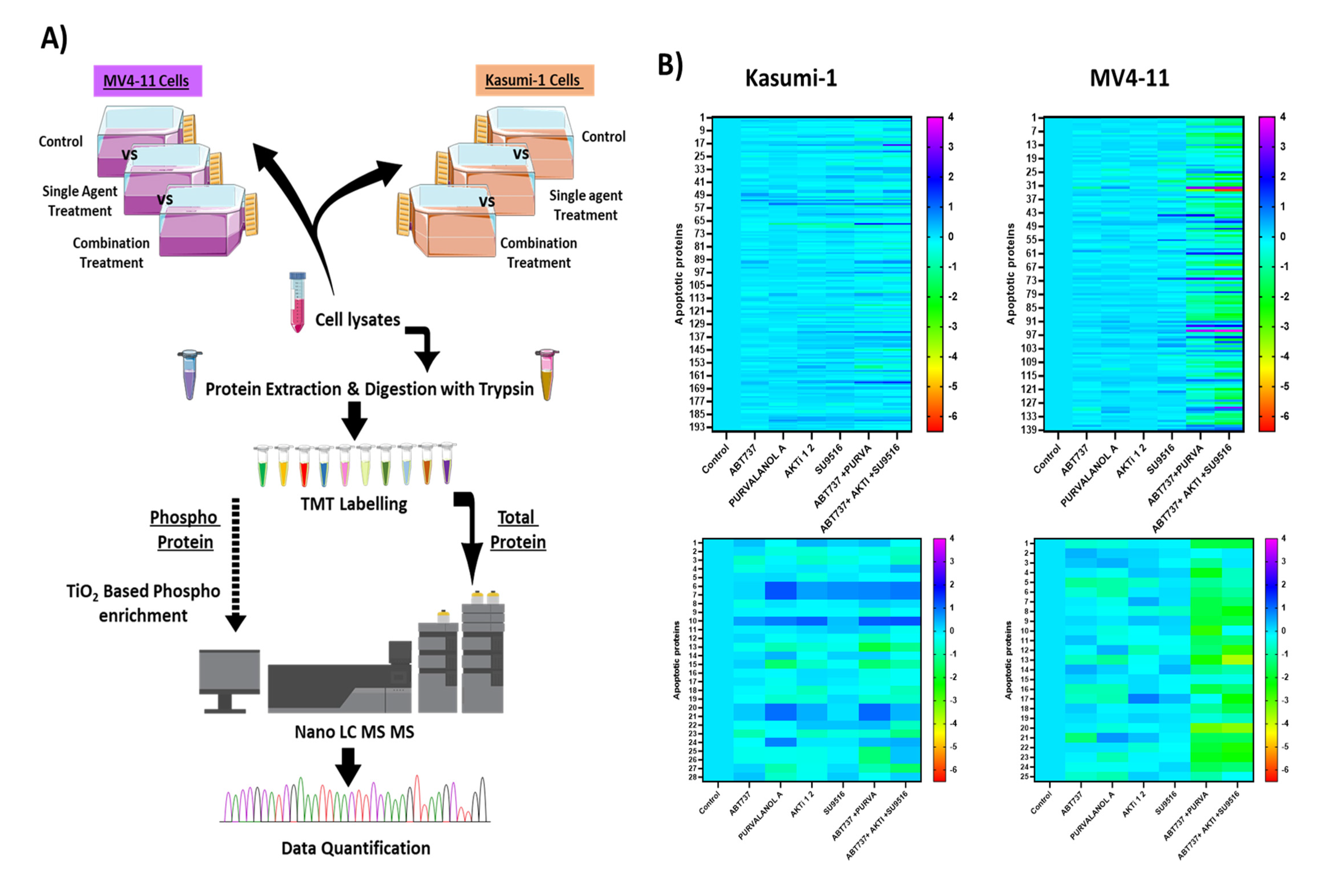
2.3. Profiling the Apoptotic Drug Combination Responsive Phosphoproteome in AML Cell Lines
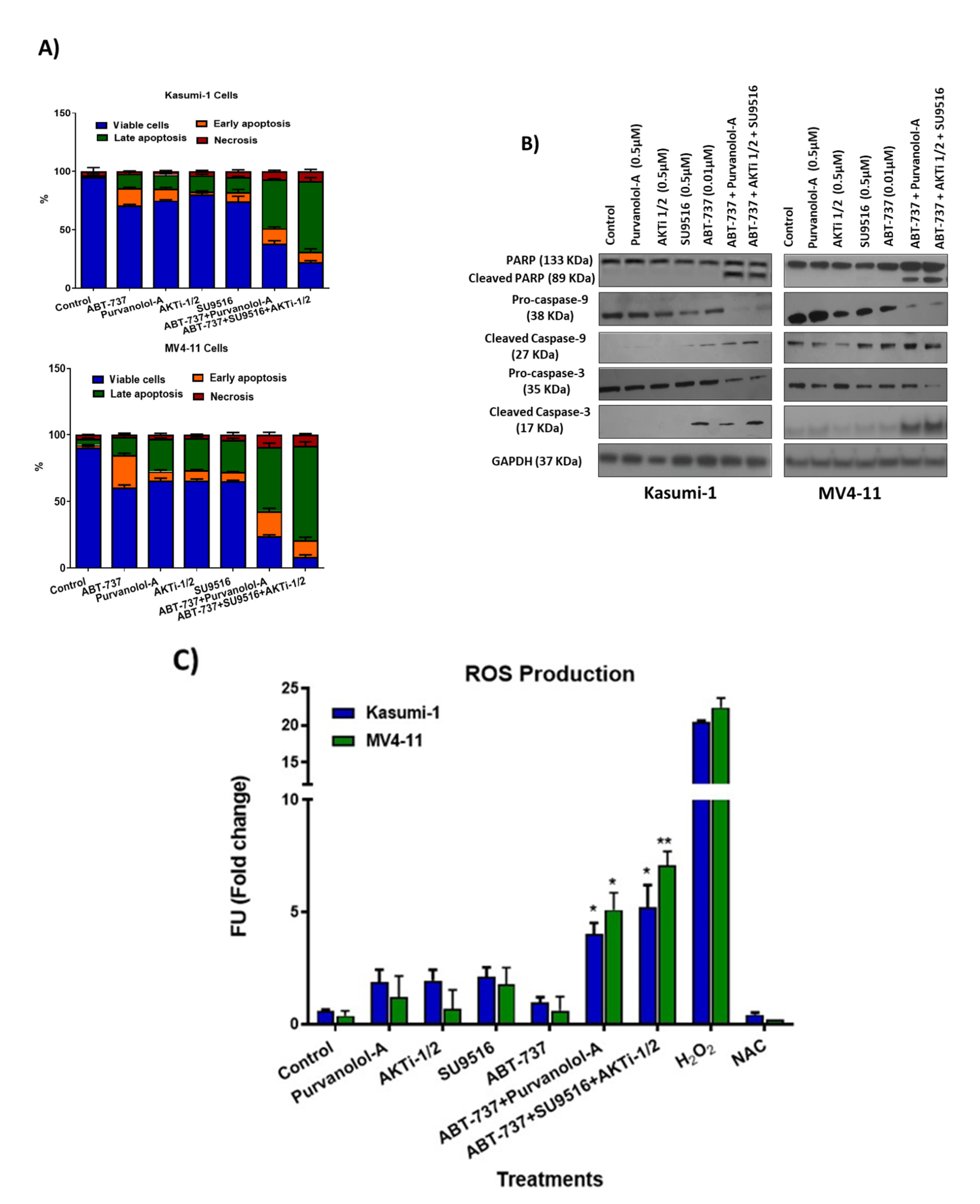
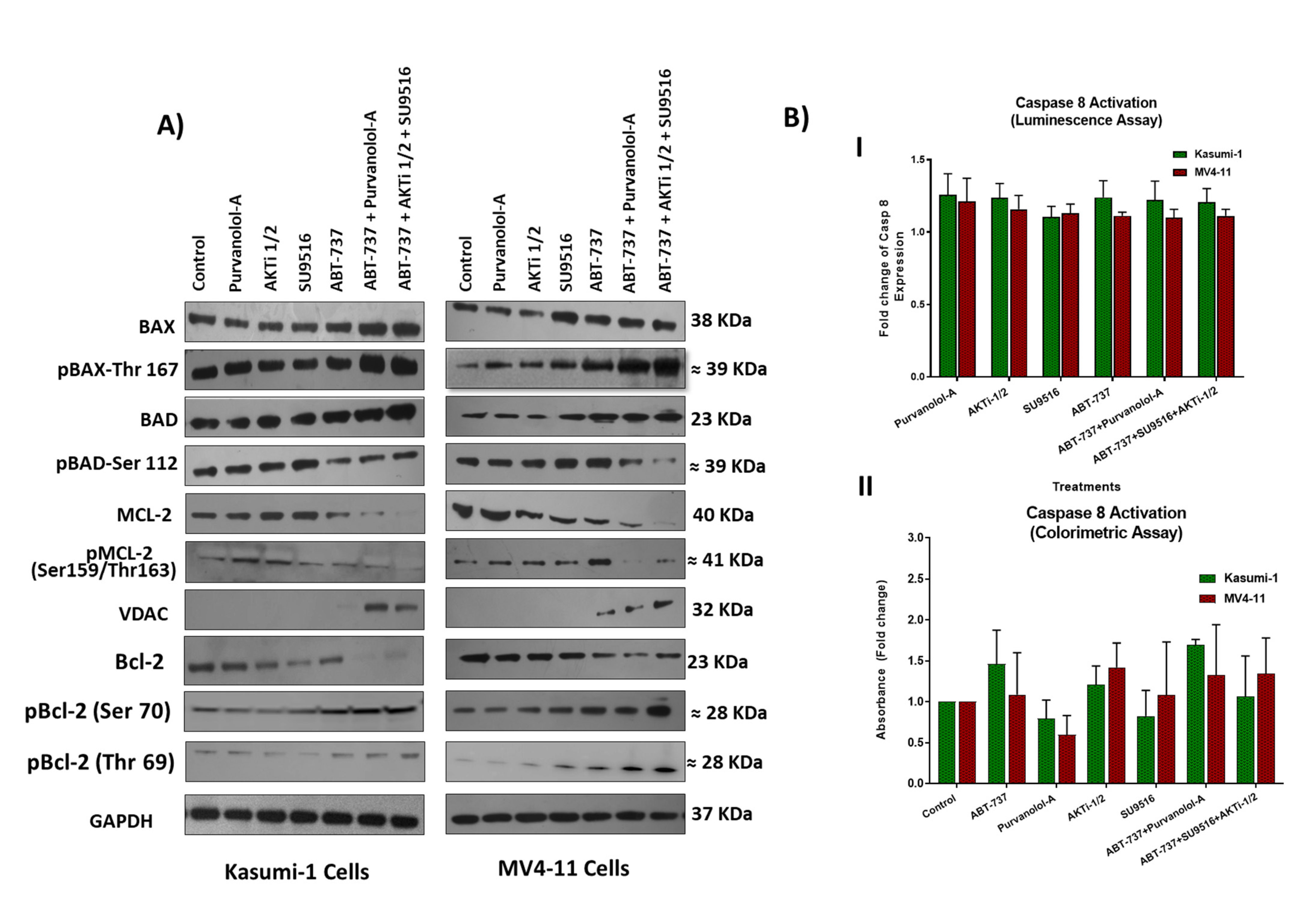
2.4. Combination Treatments Induced Cell Death through Phosphorylation of Apoptotic Proteins
2.5. Combination Therapies Induce Phosphorylation of Anti-Apoptotic Bcl-2 That Drives Apoptosis
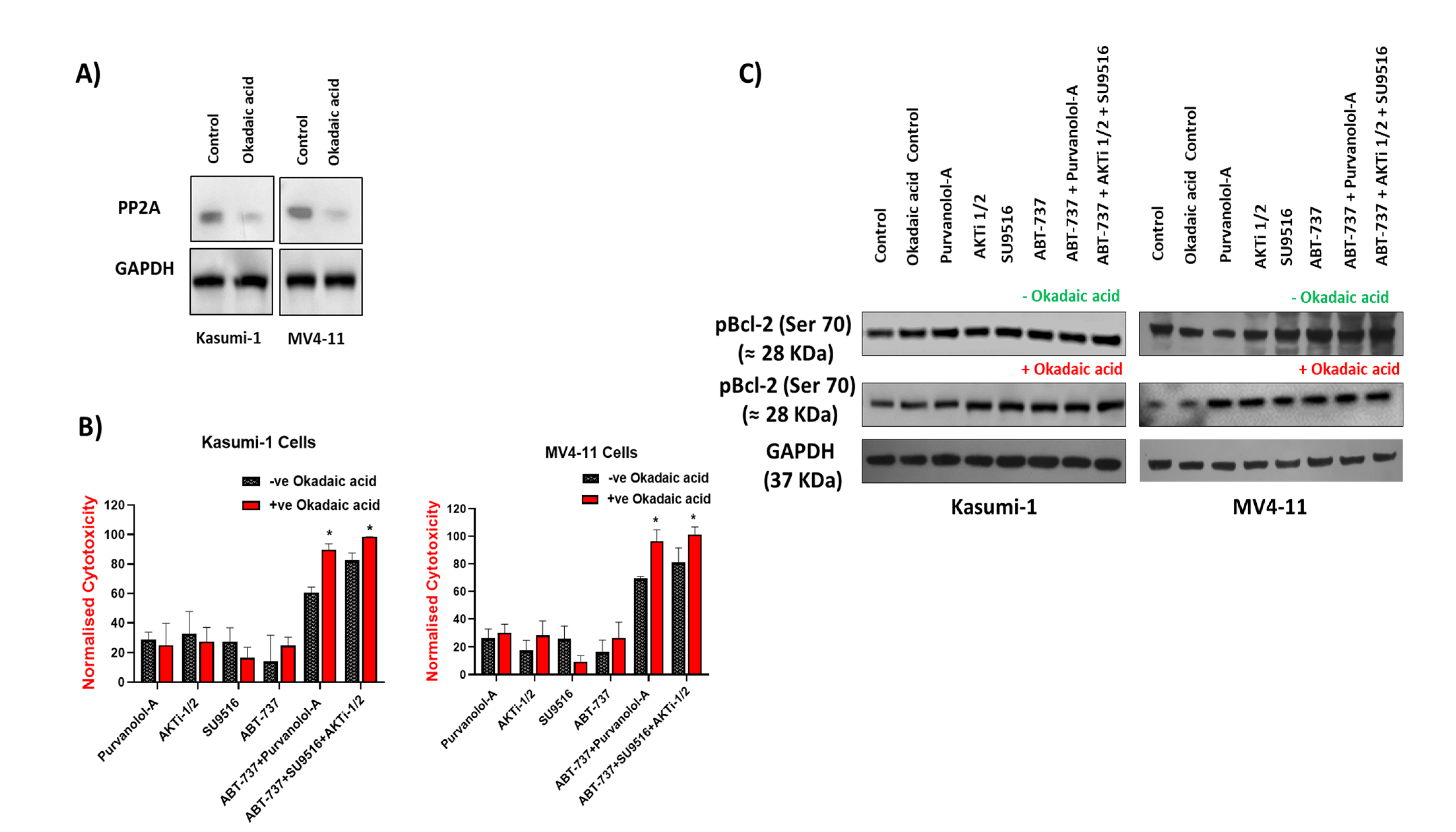
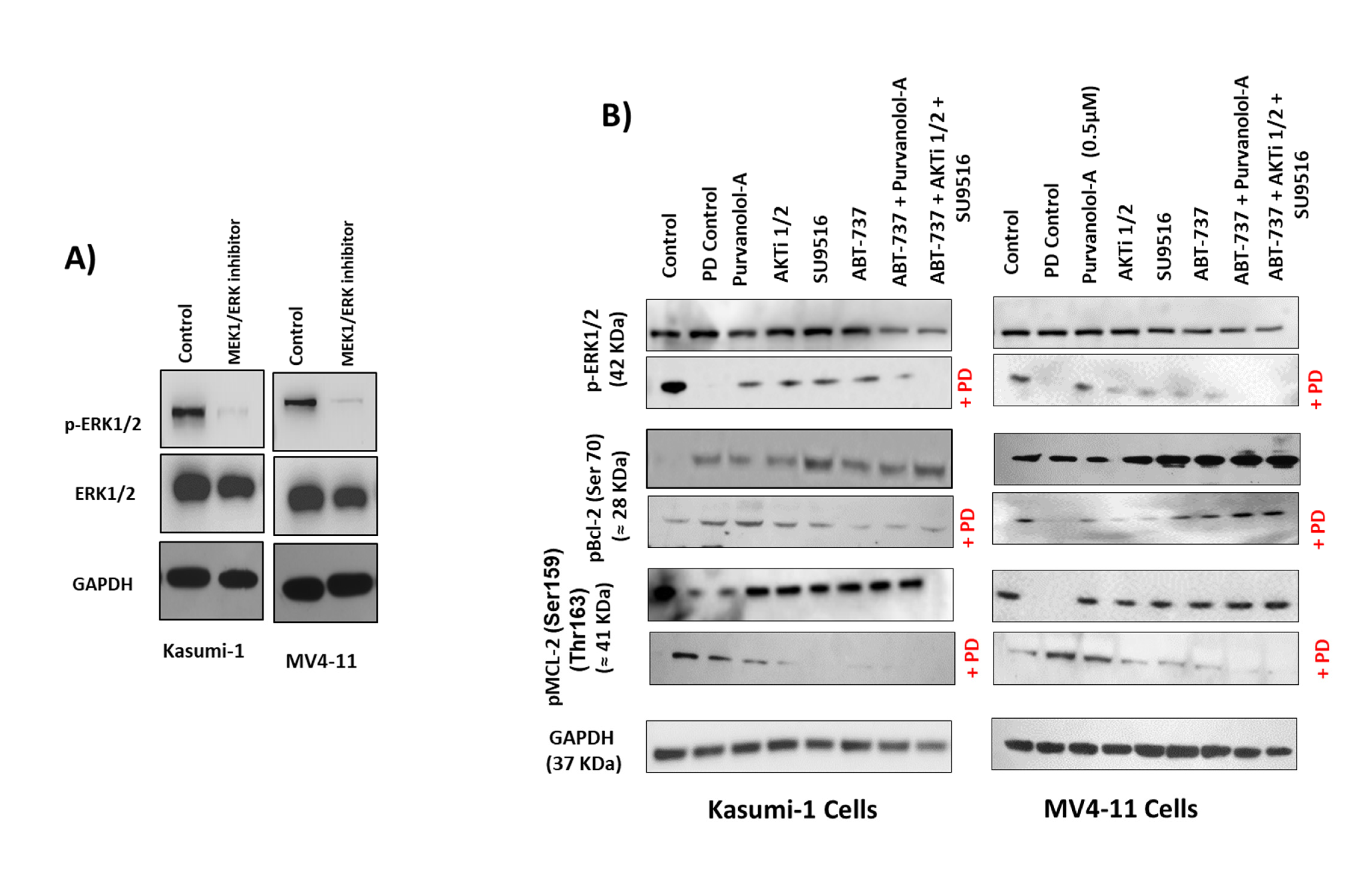
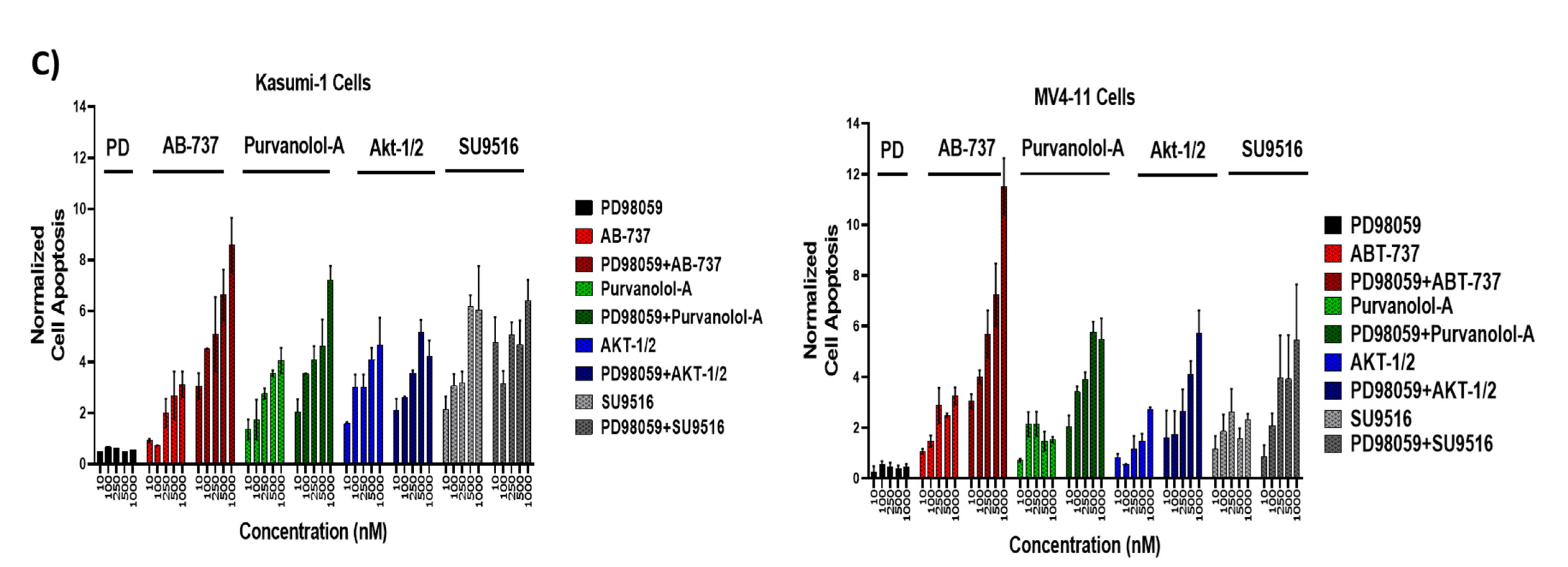
2.6. Bcl-2 Phosphorylation and Modulation of Sensitivity to Apoptotic Treatments Was Regulated by Extracellular-Signal-Regulated Kinase (ERK) but Not PP2A Phosphatase in AML Cells
3. Materials and Methods
3.1. Cell Culture
3.2. Reagents and Screening Assay
3.3. Western Blot Analysis
3.4. Annexin V Flow Cytometry
3.5. Reactive Oxygen Species Detection
3.6. Caspase-8 Activity Assays
3.7. Caspase-Glo™ Assay
3.8. Proteomic Analysis
3.9. Nano-LC–Mass Spectrometry
3.10. Proteomic Data Analysis
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kuhlen, M.; Klusmann, J.-H.; Hoell, J.I. Molecular Approaches to Treating Pediatric Leukemias. Front. Pediatr. 2019, 7, 368. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Zhang, Y.-Z.; Yan, C.-H.; Wang, Y.; Xu, L.-P.; Zhang, X.-H.; Zhang, L.-P.; Huang, X.-J.; Cheng, Y.-F. Outcomes of allogeneic haematopoietic stem cell transplantation for paediatric patients with MLL-rearranged acute myeloid leukaemia. BMC Cancer 2022, 22, 896. [Google Scholar] [CrossRef] [PubMed]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.; Marra, M.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022. [Google Scholar] [CrossRef] [Green Version]
- Ball, S.; Borthakur, G. Apoptosis targeted therapies in acute myeloid leukemia: An update. Expert Rev. Hematol. 2020, 13, 1373–1386. [Google Scholar] [CrossRef]
- Guerra, V.A.; DiNardo, C.; Konopleva, M. Venetoclax-based Therapies for Acute Myeloid Leukemia. Best Pract. Res. Clin. Haematol. 2019, 32, 145. [Google Scholar] [CrossRef] [PubMed]
- Othman, T.A.; Azenkot, T.; Moskoff, B.N.; Tenold, M.E.; Jonas, B.A. Drug Evaluation Venetoclax-based combinations for the treatment of newly diagnosed acute myeloid leukemia. Future Oncol. 2021, 17, 2989–3005. [Google Scholar] [CrossRef]
- Therapeutically Applicable Research to Generate Effective Treatments (TARGET). Available online: https://ocg.cancer.gov/programs/target (accessed on 12 November 2022).
- Lappin, K.M.; Davis, L.; Matchett, K.B.; Ge, Y.; Mills, K.I.; Blayney, J.K. A compound combination screening approach with potential to identify new treatment options for paediatric acute myeloid leukaemia. Sci. Rep. 2020, 10, 18514. [Google Scholar] [CrossRef]
- Hansen, E.; Quivoron, C.; Straley, K.; Lemieux, R.M.; Popovici-Muller, J.; Sadrzadeh, H.; Fathi, A.T.; Gliser, C.; David, M.; Saada, V.; et al. AG-120, an Oral, Selective, First-in-Class, Potent Inhibitor of Mutant IDH1, Reduces Intracellular 2HG and Induces Cellular Differentiation in TF-1 R132H Cells and Primary Human IDH1 Mutant AML Patient Samples Treated Ex Vivo. Blood 2014, 124, 3734. [Google Scholar] [CrossRef]
- Stein, E.M.; Altman, J.K.; Collins, R.; DeAngelo, D.J.; Fathi, A.T.; Flinn, I.; Frankel, A.; Levine, R.L.; Medeiros, B.C.; Patel, M.; et al. AG-221, an Oral, Selective, First-in-Class, Potent Inhibitor of the IDH2 Mutant Metabolic Enzyme, Induces Durable Remissions in a Phase I Study in Patients with IDH2 Mutation Positive Advanced Hematologic Malignancies. Blood 2014, 124, 115. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Analysis of combined drug effects: A new look at a very old problem. Trends Pharmacol. Sci. 1983, 4, 450–454. [Google Scholar] [CrossRef]
- Erba, H.P. Finding the optimal combination therapy for the treatment of newly diagnosed AML in older patients unfit for intensive therapy. Leuk. Res. 2015, 39, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Jin, F.; Wu, W.; Kumar, S.K. Cell cycle regulation and hematologic malignancies. Blood Sci. 2019, 1, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Shao, X.; Jing, H.; Liu, Y.; Qi, X.; Cao, J.; Chen, Y.; Xiang, S.; Song, H.; Hu, R.; et al. Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood 2018, 131, 2698–2711. [Google Scholar] [CrossRef] [PubMed]
- Tibes, R.; Bogenberger, J.M. Transcriptional Silencing of MCL-1 Through Cyclin-Dependent Kinase Inhibition in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1205. [Google Scholar] [CrossRef] [Green Version]
- Aasebø, E.; Berven, F.S.; Hovland, R.; Døskeland, S.O.; Bruserud, Ø.; Selheim, F.; Hernandez-Valladares, M. The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers 2020, 12, 1466. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Brenner, C.; Magrì, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 1, 108. [Google Scholar]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.; Hay, N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, L.; Cai, T.; Kuruvilla, V.M.; Cavazos, A.; Ma, H.; Battula, V.L.; Baran, N.; Harutyunyan, K.; Andreeff, M.; et al. BCL-2 Antagonist ABT-199 Combined with Complex I Inhibitor IACS-010759 Blocks Mitochondrial Respiration and Facilitates Anti-Leukemia Efficacy in Pre-Clinical AML Models. Blood 2017, 130 (Suppl. 1), 1362. [Google Scholar]
- Saletti, R.; Reina, S.; Pittalà, M.G.; Magrì, A.; Cunsolo, V.; Foti, S.; De Pinto, V. Post-translational modifications of VDAC1 and VDAC2 cysteines from rat liver mitochondria. Biochim. Biophys. Acta-Bioenerg. 2018, 1859, 806–816. [Google Scholar] [CrossRef]
- Wei, Y.; Cao, Y.; Sun, R.; Cheng, L.; Xiong, X.; Jin, X.; He, X.; Lu, W.; Zhao, M. Targeting Bcl-2 Proteins in Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 2137. [Google Scholar] [CrossRef]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 2004, 279, 21085–21095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Sun, S.Y.; Khuri, F.; Curran, W.J.; Deng, X. Mono- or Double-Site Phosphorylation Distinctly Regulates the Proapoptotic Function of Bax. PLoS ONE 2010, 5, e13393. [Google Scholar] [CrossRef] [PubMed]
- Kulsoom, B.; Shamsi, T.S.; Afsar, N.A.; Memon, Z.; Ahmed, N.; Hasnain, S.N. Bax, Bcl-2, and Bax/Bcl-2 as prognostic markers in acute myeloid leukemia: Are we ready for Bcl-2-directed therapy? Cancer Manag. Res. 2018, 10, 403. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical Role for Mitochondrial Oxidative Phosphorylation in the Activation of Tumor Suppressors Bax and Bak. JNCI J. Natl. Cancer Inst. 2006, 98, 1462–1473. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J. 2022, 41, e108690. [Google Scholar] [CrossRef]
- Papa, V.; Tazzari, P.L.; Chiarini, F.; Cappellini, A.; Ricci, F.; Billi, A.M.; Evangelisti, C.; Ottaviani, E.; Martinelli, G.; Testoni, N.; et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia 2007, 22, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Estruch, M.; Reckzeh, K.; Vittori, C.; Centio, A.; Ali, M.; Engelhard, S.; Zhao, L.; Won, K.J.; Liu, P.; Porse, B.T.; et al. Targeted inhibition of cooperative mutation- and therapy-induced AKT activation in AML effectively enhances response to chemotherapy. Leukemia 2021, 35, 2030–2042. [Google Scholar] [CrossRef]
- Estruch, M.; Vittori, C.; Muñoz-Montesinos, T.; Reckzeh, K.; Theilgaard-Mönch, K. Novel Combination Treatments for AML. J. Cell Immunol. 2021, 3, 240–245. [Google Scholar]
- Darici, S.; Alkhaldi, H.; Horne, G.; Jørgensen, H.G.; Marmiroli, S.; Huang, X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. [Google Scholar] [CrossRef]
- Chen, S.; Dai, Y.; Pei, X.Y.; Myers, J.; Wang, L.; Kramer, L.B.; Garnett, M.; Schwartz, D.M.; Su, F.; Simmons, G.L.; et al. Therapeutics, Targets, and Chemical Biology CDK Inhibitors Upregulate BH3-Only Proteins to Sensitize Human Myeloma Cells to BH3 Mimetic Therapies. Cancer Res. 2012, 72, 4225–4237. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef] [Green Version]
- Kevlicius, L.; Cepulyte, R.; Vasilevska, D.; Griskevicius, L.; Zucenka, A. Venetoclax-based regimens in combination with trametinib for RAS-mutated relapsed or refractory myeloid malignancies. Bone Marrow Transplant. 2022, 57, 1034–1037. [Google Scholar] [CrossRef]
- Van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazumder, S.; Choudhary, G.S.; Al-Harbi, S.; Almasan, A. Mcl-1 Phosphorylation Defines ABT-737 Resistance That Can Be Overcome by Increased NOXA Expression in Leukemic B cells. Cancer Res. 2012, 72, 3069–3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, P.P.; Deng, X.; May, W.S. REVIEW Phosphorylation of Bcl2 and regulation of apoptosis [Internet]. Leukemia 2001, 15, 515–522. Available online: https://www.nature.com/leu/ (accessed on 12 January 2023). [CrossRef] [Green Version]
- Shitashige, M.; Toi, M.; Yano, T.; Shibata, M.; Matsuo, Y.; Shibasaki, F. Dissociation of Bax from a Bcl-2/Bax Heterodimer Triggered by Phosphorylation of Serine 70 of Bcl-2. J. Biochem. 2001, 130, 741–748. Available online: https://academic.oup.com/jb/article/130/6/741/798410 (accessed on 15 January 2023). [CrossRef] [PubMed]
- Arriazu, E.; Pippa, R.; Odero, M.D. Protein Phosphatase 2A as a Therapeutic Target in Acute Myeloid Leukemia. Front. Oncol. 2016, 6, 78. [Google Scholar] [CrossRef] [Green Version]
- Soldovieri, M.V. Okadaic acid. xPharm. Compr. Pharmacol. Ref. 2009, 1–6. [Google Scholar]
- Lange, B.; Valtieri, M.; Santoli, D.; Caracciolo, D.; Mavilio, F.; Gemperlein, I.; Griffin, C.; Emanuel, B.; Finan, J.; Nowell, P. Growth Factor Requirements of Childhood Acute Leukemia: Establishment of GM-CSF-Dependent Cell Lines. Blood 1987, 70, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asou, H.; Tashiro, S.; Hamamoto, K.; Otsuji, A.; Kita, K.; Kamada, N. Establishment of a Human Acute Myeloid Leukemia Cell Line (Kasumi-1) With 8;21 Chromosome Translocation. Blood 1991, 77, 2031–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, X.; Wang, G.; Wang, Y.; Caldwell, J.T.; Edwards, H.; Xie, C.; Taub, J.W.; Li, C.; Lin, H.; Ge, Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 2014, 28, 1557–1560. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phosphoproteome and Proteome Dataset | Kasumi-1 Cells | MV4-11 Cells |
|---|---|---|
| Unique phosphosites | 1646 | 1349 |
| Total: 2468 | Overlap: 527 | |
| Unique phosphopeptides | 1464 | 1186 |
| Total: 2022 | Overlap: 628 | |
| Unique phosphoproteins | 897 | 706 |
| Unique phosphopeptides w/matching prot | 1530 | 1267 |
| Proteins (proteomic analysis) | 7554 | 6978 |
| Total: 8901 | Overlap: 5631 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.A.; Cairns, L.V.; Clarke, K.M.; Blayney, J.K.; Lappin, K.M.; Mills, K.I. Combination Therapies Targeting Apoptosis in Paediatric AML: Understanding the Molecular Mechanisms of AML Treatments Using Phosphoproteomics. Int. J. Mol. Sci. 2023, 24, 5717. https://doi.org/10.3390/ijms24065717
Ali AA, Cairns LV, Clarke KM, Blayney JK, Lappin KM, Mills KI. Combination Therapies Targeting Apoptosis in Paediatric AML: Understanding the Molecular Mechanisms of AML Treatments Using Phosphoproteomics. International Journal of Molecular Sciences. 2023; 24(6):5717. https://doi.org/10.3390/ijms24065717
Chicago/Turabian StyleAli, Ahlam A., Lauren V. Cairns, Kathryn M. Clarke, Jaine K. Blayney, Katrina M. Lappin, and Ken I. Mills. 2023. "Combination Therapies Targeting Apoptosis in Paediatric AML: Understanding the Molecular Mechanisms of AML Treatments Using Phosphoproteomics" International Journal of Molecular Sciences 24, no. 6: 5717. https://doi.org/10.3390/ijms24065717
APA StyleAli, A. A., Cairns, L. V., Clarke, K. M., Blayney, J. K., Lappin, K. M., & Mills, K. I. (2023). Combination Therapies Targeting Apoptosis in Paediatric AML: Understanding the Molecular Mechanisms of AML Treatments Using Phosphoproteomics. International Journal of Molecular Sciences, 24(6), 5717. https://doi.org/10.3390/ijms24065717