
Mouse Models of Gestational Diabetes Mellitus and Its Subtypes: Recent Insights and Pitfalls



Abstract
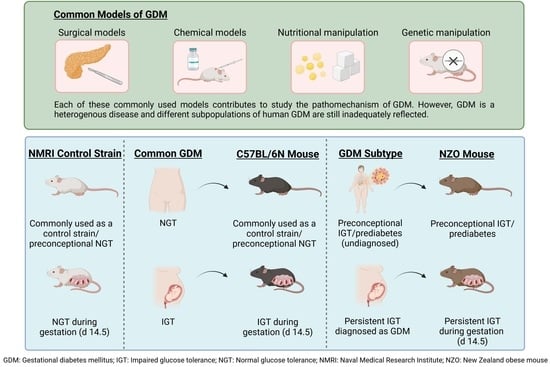
:
1. Introduction
2. Genetic Risk Factors of GDM
3. Subtypes of GDM
4. Conventional Mouse Models of GDM
5. The New Zealand Obese (NZO) Mouse—A Model of a Common Subpopulation of Human GDM
6. The C57BL/6N Mouse—A Model of Human GDM
7. Choosing the Right Control for Metabolic Studies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association Professional Practice Committee. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S17–S38. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, H.D.; Catalano, P.; Zhang, C.; Desoye, G.; Mathiesen, E.R.; Damm, P. Gestational diabetes mellitus. Nat. Rev. Dis. Prim. 2019, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Du, S.; Sun, D.; Li, X.; Heianza, Y.; Hu, G.; Sun, L.; Pei, X.; Shang, X.; Qi, L. Prevalence and Trends in Gestational Diabetes Mellitus Among Women in the United States, 2006–2017: A Population-Based Study. Front. Endocrinol. 2022, 13, 868094. [Google Scholar] [CrossRef] [PubMed]
- International Association of Diabetes and Pregnancy Study Groups Consensus Panel. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 2010, 33, 676–682. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes—2019. Diabetes Care 2019, 42, S13–S28. [Google Scholar] [CrossRef] [Green Version]
- Blumer, I.; Hadar, E.; Hadden, D.R.; Jovanovič, L.; Mestman, J.H.; Murad, M.H.; Yogev, Y. Diabetes and pregnancy: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2013, 98, 4227–4249. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hou, W.; Meng, X.; Zhao, W.; Pan, J.; Tang, J.; Huang, Y.; Tao, M.; Liu, F. Heterogeneity of insulin resistance and beta cell dysfunction in gestational diabetes mellitus: A prospective cohort study of perinatal outcomes. J. Transl. Med. 2018, 16, 289. [Google Scholar] [CrossRef]
- Buchanan, T.A.; Xiang, A.H.; Page, K.A. Gestational Diabetes Mellitus: Risks and Management during and after Pregnancy. Nat. Rev. Endocrinol. 2012, 8, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.M.; Kim, T.; Lim, S.; Choi, S.H.; Shin, H.; Lee, H.; Park, K.S.; Jang, H.C. Type 2 diabetes-associated genetic variants discovered in the recent genome-wide association studies are related to gestational diabetes mellitus in the Korean population. Diabetologia 2009, 52, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Huopio, H.; Cederberg, H.; Vangipurapu, J.; Hakkarainen, H.; Pääkkönen, M.; Kuulasmaa, T.; Heinonen, S.; Laakso, M. Association of risk variants for type 2 diabetes and hyperglycemia with gestational diabetes. Eur. J. Endocrinol. 2013, 169, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Robitaille, J.; Grant, A.M. The genetics of gestational diabetes mellitus: Evidence for relationship with type 2 diabetes mellitus. Genet. Med. 2008, 10, 240. [Google Scholar] [CrossRef] [Green Version]
- IDF. IDF Diabetes Atlas 10th Edition. Available online: https://diabetesatlas.org/idfawp/resource-files/2021/07/IDF_Atlas_10th_Edition_2021.pdf (accessed on 29 November 2022).
- Xiang, A.H.; Kjos, S.L.; Takayanagi, M.; Trigo, E.; Buchanan, T.A. Detailed Physiological Characterization of the Development of Type 2 Diabetes in Hispanic Women With Prior Gestational Diabetes Mellitus. Diabetes 2010, 59, 2625–2630. [Google Scholar] [CrossRef] [Green Version]
- Dall, T.M.; Yang, W.; Halder, P.; Pang, B.; Massoudi, M.; Wintfeld, N.; Semilla, A.P.; Franz, J.; Hogan, P.F. The economic burden of elevated blood glucose levels in 2012: Diagnosed and undiagnosed diabetes, gestational diabetes mellitus, and prediabetes. Diabetes Care 2014, 37, 3172–3179. [Google Scholar] [CrossRef] [Green Version]
- Wagner, R.; Heni, M.; Tabák, A.G.; Machann, J.; Schick, F.; Randrianarisoa, E.; de Angelis, M.H.; Birkenfeld, A.L.; Stefan, N.; Peter, A. Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat. Med. 2021, 27, 49–57. [Google Scholar] [CrossRef]
- Chatzigeorgiou, A.; Halapas, A.; Kalafatakis, K.; Kamper, E. The use of animal models in the study of diabetes mellitus. In Vivo 2009, 23, 245–258. [Google Scholar]
- Peters, L.L.; Robledo, R.F.; Bult, C.J.; Churchill, G.A.; Paigen, B.J.; Svenson, K.L. The mouse as a model for human biology: A resource guide for complex trait analysis. Nat. Rev. Genet. 2007, 8, 58–69. [Google Scholar] [CrossRef]
- Pasek, R.C.; Gannon, M. Advancements and challenges in generating accurate animal models of gestational diabetes mellitus. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, E1327–E1338. [Google Scholar] [CrossRef] [Green Version]
- Popova, P.V.; Klyushina, A.A.; Vasilyeva, L.B.; Tkachuk, A.S.; Vasukova, E.A.; Anopova, A.D.; Pustozerov, E.A.; Gorelova, I.V.; Kravchuk, E.N.; Li, O. Association of common genetic risk variants with gestational diabetes mellitus and their role in GDM prediction. Front. Endocrinol. 2021, 12, 628582. [Google Scholar] [CrossRef]
- Powe, C.E.; Kwak, S.H. Genetic studies of gestational diabetes and glucose metabolism in pregnancy. Curr. Diabetes Rep. 2020, 20, 69. [Google Scholar] [CrossRef]
- Mao, H.; Li, Q.; Gao, S. Meta-Analysis of the Relationship between Common Type 2 Diabetes Risk Gene Variants with Gestational Diabetes Mellitus. PLoS ONE 2012, 7, e45882. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Cui, L.; Tam, W.H.; Ma, R.C.W.; Wang, C.C. Genetic variants associated with gestational diabetes mellitus: A meta-analysis and subgroup analysis. Sci. Rep. 2016, 6, 30539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jääskeläinen, T.; Klemetti, M.M. Genetic Risk Factors and Gene–Lifestyle Interactions in Gestational Diabetes. Nutrients 2022, 14, 4799. [Google Scholar] [CrossRef] [PubMed]
- Angueira, A.R.; Ludvik, A.E.; Reddy, T.E.; Wicksteed, B.; Lowe, W.L.; Layden, B.T. New insights into gestational glucose metabolism: Lessons learned from 21st century approaches. Diabetes 2015, 64, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, W.L.; Scholtens, D.M.; Sandler, V.; Hayes, M.G. Genetics of gestational diabetes mellitus and maternal metabolism. Curr. Diabetes Rep. 2016, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Y.; Song, L.-P.; Wei, S.-D.; Wen, X.-L.; Liu, D.-B. CDK5 Regulatory Subunit-Associated Protein 1-Like 1 Gene Polymorphisms and Gestational Diabetes Mellitus Risk: A Trial Sequential Meta-Analysis of 13,306 Subjects. Front. Endocrinol. 2021, 12, 722674. [Google Scholar] [CrossRef]
- Wang, K.; Chen, Q.; Feng, Y.; Yang, H.; Wu, W.; Zhang, P.; Wang, Y.; Ko, J.; Zhao, F.; Du, W. Single nucleotide polymorphisms in CDKAL1 gene are associated with risk of gestational diabetes mellitus in Chinese population. J. Diabetes Res. 2019, 2019, 3618103. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.; Leng, J.; Li, W.; Liu, J.; Yan, X.; Yu, Z.; Hu, G.; Ma, R.C.W.; Fang, Z.; et al. The CDKAL1 rs7747752-Bile Acids Interaction Increased Risk of Gestational Diabetes Mellitus: A Nested Case-Control Study. Front. Endocrinol. 2022, 13, 808956. [Google Scholar] [CrossRef]
- Wang, Y.; Nie, M.; Li, W.; Ping, F.; Hu, Y.; Ma, L.; Gao, J.; Liu, J. Association of Six Single Nucleotide Polymorphisms with Gestational Diabetes Mellitus in a Chinese Population. PLoS ONE 2011, 6, e26953. [Google Scholar] [CrossRef] [Green Version]
- Hannou, S.A.; Wouters, K.; Paumelle, R.; Staels, B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: What have we learned from GWASs? Trends Endocrinol. Metab. 2015, 26, 176–184. [Google Scholar] [CrossRef]
- Zhang, C.; Bao, W.; Rong, Y.; Yang, H.; Bowers, K.; Yeung, E.; Kiely, M. Genetic variants and the risk of gestational diabetes mellitus: A systematic review. Hum. Reprod. Update 2013, 19, 376–390. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, L.; Qiang, P. The role of IGF2BP2, an m6A reader gene, in human metabolic diseases and cancers. Cancer Cell Int. 2021, 21, 99. [Google Scholar] [CrossRef]
- Shaat, N.; Ekelund, M.; Lernmark, Å.; Ivarsson, S.; Almgren, P.; Berntorp, K.; Groop, L. Association of the E23K polymorphism in the KCNJ11 gene with gestational diabetes mellitus. Diabetologia 2005, 48, 2544–2551. [Google Scholar] [CrossRef] [Green Version]
- Ao, D.; Wang, H.-J.; Wang, L.-F.; Song, J.-Y.; Yang, H.-X.; Wang, Y. The rs2237892 Polymorphism in KCNQ1 Influences Gestational Diabetes Mellitus and Glucose Levels: A Case-Control Study and Meta-Analysis. PLoS ONE 2015, 10, e0128901. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Qiao, B.; Zhan, Y.; Peng, W.; Chen, Z.-J.; Sun, L.; Zhang, J.; Zhao, L.; Gao, Q. Association between genetic variations in MTNR1A and MTNR1B genes and gestational diabetes mellitus in Han Chinese women. Gynecol. Obstet. Investig. 2013, 76, 221–227. [Google Scholar] [CrossRef]
- Shaat, N.; Lernmark, Å.; Karlsson, E.; Ivarsson, S.; Parikh, H.; Berntorp, K.; Groop, L. A variant in the transcription factor 7-like 2 (TCF7L2) gene is associated with an increased risk of gestational diabetes mellitus. Diabetologia 2007, 50, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.I. Gestational Diabetes May Represent Discovery of Preexisting Glucose Intolerance. Diabetes Care 1988, 11, 402–411. [Google Scholar] [CrossRef]
- Raets, L.; Beunen, K.; Benhalima, K. Screening for gestational diabetes mellitus in early pregnancy: What is the evidence? J. Clin. Med. 2021, 10, 1257. [Google Scholar] [CrossRef]
- Zhang, C.; Rawal, S.; Chong, Y.S. Risk factors for gestational diabetes: Is prevention possible? Diabetologia 2016, 59, 1385–1390. [Google Scholar] [CrossRef] [Green Version]
- Immanuel, J.; Simmons, D. Screening and Treatment for Early-Onset Gestational Diabetes Mellitus: A Systematic Review and Meta-analysis. Curr. Diabetes Rep. 2017, 17, 115. [Google Scholar] [CrossRef]
- Wexler, D.J.; Powe, C.E.; Barbour, L.A.; Buchanan, T.; Coustan, D.R.; Corcoy, R.; Damm, P.; Dunne, F.; Feig, D.S.; Ferrara, A.; et al. Research Gaps in Gestational Diabetes Mellitus: Executive Summary of a National Institute of Diabetes and Digestive and Kidney Diseases Workshop. Obstet. Gynecol. 2018, 132, 496–505. [Google Scholar] [CrossRef]
- Cheney, C.; Shragg, P.; Hollingsworth, D. Demonstration of heterogeneity in gestational diabetes by a 400-kcal breakfast meal tolerance test. Obstet. Gynecol. 1985, 65, 17–23. [Google Scholar] [PubMed]
- Powe, C.E.; Allard, C.; Battista, M.-C.; Doyon, M.; Bouchard, L.; Ecker, J.L.; Perron, P.; Florez, J.C.; Thadhani, R.; Hivert, M.-F. Heterogeneous contribution of insulin sensitivity and secretion defects to gestational diabetes mellitus. Diabetes Care 2016, 39, 1052–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am. J. Obstet. Gynecol. 1999, 180, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Huvinen, E.; Eriksson, J.G.; Koivusalo, S.B.; Grotenfelt, N.; Tiitinen, A.; Stach-Lempinen, B.; Rönö, K. Heterogeneity of gestational diabetes (GDM) and long-term risk of diabetes and metabolic syndrome: Findings from the RADIEL study follow-up. Acta Diabetol. 2018, 55, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huvinen, E.; Grotenfelt, N.E.; Eriksson, J.G.; Rönö, K.; Klemetti, M.M.; Roine, R.; Pöyhönen-Alho, M.; Tiitinen, A.; Andersson, S.; Laivuori, H.; et al. Heterogeneity of maternal characteristics and impact on gestational diabetes (GDM) risk-Implications for universal GDM screening? Ann. Med. 2016, 48, 52–58. [Google Scholar] [CrossRef]
- Benhalima, K.; Van Crombrugge, P.; Moyson, C.; Verhaeghe, J.; Vandeginste, S.; Verlaenen, H.; Vercammen, C.; Maes, T.; Dufraimont, E.; De Block, C.; et al. Characteristics and pregnancy outcomes across gestational diabetes mellitus subtypes based on insulin resistance. Diabetologia 2019, 62, 2118–2128. [Google Scholar] [CrossRef]
- Immanuel, J.; Simmons, D.; Harreiter, J.; Desoye, G.; Corcoy, R.; Adelantado, J.M.; Devlieger, R.; Lapolla, A.; Dalfra, M.G.; Bertolotto, A.; et al. Metabolic phenotypes of early gestational diabetes mellitus and their association with adverse pregnancy outcomes. Diabet. Med. 2021, 38, e14413. [Google Scholar] [CrossRef]
- Kotzaeridi, G.; Blätter, J.; Eppel, D.; Rosicky, I.; Linder, T.; Geissler, F.; Huhn, E.A.; Hösli, I.; Tura, A.; Göbl, C.S. Characteristics of gestational diabetes subtypes classified by oral glucose tolerance test values. Eur. J. Clin. Investig. 2021, 51, e13628. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kobayashi, N.; Okitsu, T.; Yong, C.; Fukazawa, T.; Ikeda, H.; Kosaka, Y.; Narushima, M.; Arata, T.; Tanaka, N. Development of a porcine model of type 1 diabetes by total pancreatectomy and establishment of a glucose tolerance evaluation method. Artif. Organs 2004, 28, 1035–1042. [Google Scholar] [CrossRef]
- Jarrett, I.; Potter, B.; Packham, A. Effects of pancreatectomy in the sheep. Aust. J. Exp. Biol. Med. Sci. 1956, 34, 133–141. [Google Scholar] [CrossRef]
- Cuthbert, F.; Ivy, A.; Isaacs, B.; Gray, J. The relation of pregnancy and lactation to extirpation diabetes in the dog. Am. J. Physiol.-Leg. Content 1936, 115, 480–496. [Google Scholar] [CrossRef]
- Gillman, J.; Gilbert, C.; Epstein, E.; Allan, J. Endocrine control of blood sugar, lipaemia, and ketonaemia in diabetic baboons. Br. Med. J. 1958, 2, 1260. [Google Scholar] [CrossRef] [Green Version]
- De Sousa, R.A.L. Animal models of gestational diabetes: Characteristics and consequences to the brain and behavior of the offspring. Metab. Brain Dis. 2021, 36, 199–204. [Google Scholar] [CrossRef]
- Yamashita, H.; Shao, J.; Ishizuka, T.; Klepcyk, P.J.; Muhlenkamp, P.; Qiao, L.; Hoggard, N.; Friedman, J.E. Leptin administration prevents spontaneous gestational diabetes in heterozygous Leprdb/+ mice: Effects on placental leptin and fetal growth. Endocrinology 2001, 142, 2888–2897. [Google Scholar] [CrossRef]
- Yamashita, H.; Shao, J.; Qiao, L.; Pagliassotti, M.; Friedman, J.E. Effect of spontaneous gestational diabetes on fetal and postnatal hepatic insulin resistance in Lepr db/+ mice. Pediatr. Res. 2003, 53, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, R.C.; Amankwah, K.S.; Dunaway, G.; Maroun, L.; Arbuthnot, J.; Roddick, J., Jr. An animal model of gestational diabetes. Am. J. Obstet. Gynecol. 1981, 141, 479–482. [Google Scholar] [CrossRef]
- Plows, J.F.; Yu, X.; Broadhurst, R.; Vickers, M.H.; Tong, C.; Zhang, H.; Qi, H.; Stanley, J.L.; Baker, P.N. Absence of a gestational diabetes phenotype in the LepRdb/+ mouse is independent of control strain, diet, misty allele, or parity. Sci. Rep. 2017, 7, 45130. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Snider, F.; Cross, J.C. Prolactin receptor is required for normal glucose homeostasis and modulation of β-cell mass during pregnancy. Endocrinology 2008, 150, 1618–1626. [Google Scholar] [CrossRef] [Green Version]
- Freemark, M.; Avril, I.; Fleenor, D.; Driscoll, P.; Petro, A.; Opara, E.; Kendall, W.; Oden, J.; Bridges, S.; Binart, N. Targeted deletion of the PRL receptor: Effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002, 143, 1378–1385. [Google Scholar] [CrossRef]
- Bole-Feysot, C.; Goffin, V.; Edery, M.; Binart, N.; Kelly, P.A. Prolactin (PRL) and its receptor: Actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr. Rev. 1998, 19, 225–268. [Google Scholar] [CrossRef]
- Baeyens, L.; Hindi, S.; Sorenson, R.L.; German, M.S. β-Cell adaptation in pregnancy. Diabetes Obes. Metab. 2016, 18, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat. Med. 2010, 16, 804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ackermann, A.M.; Gusarova, G.A.; Lowe, D.; Feng, X.; Kopsombut, U.G.; Costa, R.H.; Gannon, M. The FoxM1 transcription factor is required to maintain pancreatic β-cell mass. Mol. Endocrinol. 2006, 20, 1853–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhang, J.; Pope, C.F.; Crawford, L.A.; Vasavada, R.C.; Jagasia, S.M.; Gannon, M. Gestational Diabetes Mellitus Resulting From Impaired β-Cell Compensation in the Absence of FoxM1, a Novel Downstream Effector of Placental Lactogen. Diabetes 2010, 59, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Kluge, R.; Scherneck, S.; Schürmann, A.; Joost, H.-G. Pathophysiology and genetics of obesity and diabetes in the New Zealand obese mouse: A model of the human metabolic syndrome. In Animal Models in Diabetes Research; Springer: Berlin/Heidelberg, Germany, 2012; pp. 59–73. [Google Scholar]
- Bielschowsky, M.; Goodall, C. Origin of inbred NZ mouse strains. Cancer Res. 1970, 30, 834–836. [Google Scholar]
- Igel, M.; Becker, W.; Herberg, L.; Joost, H.-G. Hyperleptinemia, leptin resistance, and polymorphic leptin receptor in the New Zealand obese mouse. Endocrinology 1997, 138, 4234–4239. [Google Scholar] [CrossRef]
- Jürgens, H.S.; Schürmann, A.; Kluge, R.; Ortmann, S.; Klaus, S.; Joost, H.-G.; Tschöp, M.H. Hyperphagia, lower body temperature, and reduced running wheel activity precede development of morbid obesity in New Zealand obese mice. Physiol. Genom. 2006, 25, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Joost, H.-G. Pathogenesis, risk assessment and prevention of type 2 diabetes mellitus. Obes. Facts 2008, 1, 128–137. [Google Scholar] [CrossRef]
- Scherneck, S.; Nestler, M.; Vogel, H.; Blüher, M.; Block, M.-D.; Diaz, M.B.; Herzig, S.; Schulz, N.; Teichert, M.; Tischer, S.; et al. Positional Cloning of Zinc Finger Domain Transcription Factor Zfp69, a Candidate Gene for Obesity-Associated Diabetes Contributed by Mouse Locus Nidd/SJL. PLoS Genet. 2009, 5, e1000541. [Google Scholar] [CrossRef] [Green Version]
- Chadt, A.; Leicht, K.; Deshmukh, A.; Jiang, L.Q.; Scherneck, S.; Bernhardt, U.; Dreja, T.; Vogel, H.; Schmolz, K.; Kluge, R.; et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat. Genet. 2008, 40, 1354. [Google Scholar] [CrossRef]
- Kanasaki, K.; Koya, D. Biology of obesity: Lessons from animal models of obesity. J. Biomed. Biotechnol. 2011, 2011, 197636. [Google Scholar] [CrossRef] [Green Version]
- Leiter, E.H. Selecting the “right” mouse model for metabolic syndrome and type 2 diabetes research. In Type 2 Diabetes; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1–17. [Google Scholar]
- Le May, C.; Chu, K.; Hu, M.; Ortega, C.S.; Simpson, E.R.; Korach, K.S.; Tsai, M.-J.; Mauvais-Jarvis, F. Estrogens protect pancreatic β-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9232–9237. [Google Scholar] [CrossRef] [Green Version]
- Louet, J.-F.; LeMay, C.; Mauvais-Jarvis, F. Antidiabetic actions of estrogen: Insight from human and genetic mouse models. Curr. Atheroscler. Rep. 2004, 6, 180–185. [Google Scholar] [CrossRef]
- Lubura, M.; Hesse, D.; Kraemer, M.; Hallahan, N.; Schupp, M.; von Loffelholz, C.; Kriebel, J.; Rudovich, N.; Pfeiffer, A.; John, C.; et al. Diabetes prevalence in NZO females depends on estrogen action on liver fat content. Am. J. Physiol.-Endocrinol. Metab. 2015, 309, E968–E980. [Google Scholar] [CrossRef] [Green Version]
- Vogel, H.; Mirhashemi, F.; Liehl, B.; Taugner, F.; Kluth, O.; Kluge, R.; Joost, H.G.; Schürmann, A. Estrogen deficiency aggravates insulin resistance and induces beta-cell loss and diabetes in female New Zealand obese mice. Horm. Metab. Res. 2013, 45, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Grupe, K.; Asuaje Pfeifer, M.; Dannehl, F.; Liebmann, M.; Rustenbeck, I.; Schurmann, A.; Scherneck, S. Metabolic changes during pregnancy in glucose-intolerant NZO mice: A polygenic model with prediabetic metabolism. Physiol. Rep. 2020, 8, e14417. [Google Scholar] [CrossRef]
- Almaça, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; Berggren, P.-O.; Caicedo, A. Human beta cells produce and release serotonin to inhibit glucagon secretion from alpha cells. Cell Rep. 2016, 17, 3281–3291. [Google Scholar] [CrossRef] [Green Version]
- Goyvaerts, L.; Schraenen, A.; Schuit, F. Serotonin competence of mouse beta cells during pregnancy. Diabetologia 2016, 59, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Schraenen, A.; Lemaire, K.; de Faudeur, G.; Hendrickx, N.; Granvik, M.; Van Lommel, L.; Mallet, J.; Vodjdani, G.; Gilon, P.; Binart, N.; et al. Placental lactogens induce serotonin biosynthesis in a subset of mouse beta cells during pregnancy. Diabetologia 2010, 53, 2589–2599. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhu, Y.; Zhou, W.; Gao, L.; Yuan, L.; Han, X. Serotonin receptor 2C and insulin secretion. PLoS ONE 2013, 8, e54250. [Google Scholar] [CrossRef]
- Cataldo, L.R.; Mizgier, M.L.; Bravo Sagua, R.; Jana, F.; Cardenas, C.; Llanos, P.; Busso, D.; Olmos, P.; Galgani, J.E.; Santos, J.L. Prolonged activation of the Htr2b serotonin receptor impairs glucose stimulated insulin secretion and mitochondrial function in MIN6 cells. PLoS ONE 2017, 12, e0170213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulmann, N.; Grohmann, M.; Voigt, J.-P.; Bert, B.; Vowinckel, J.; Bader, M.; Skelin, M.; Jevšek, M.; Fink, H.; Rupnik, M. Intracellular serotonin modulates insulin secretion from pancreatic β-cells by protein serotonylation. PLoS Biol. 2009, 7, e1000229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asuaje Pfeifer, M.; Liebmann, M.; Beuerle, T.; Grupe, K.; Scherneck, S. Role of Serotonin (5-HT) in GDM Prediction Considering Islet and Liver Interplay in Prediabetic Mice during Gestation. Int. J. Mol. Sci. 2022, 23, 6434. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Alonso, A.; Alvarez, N.; Diaz, F.; Martinez, M.; Fernandez, S.; Patterson, A. Role of 17beta-estradiol and/or progesterone on insulin sensitivity in the rat: Implications during pregnancy. J. Endocrinol. 2000, 166, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebmann, M.; Asuaje Pfeifer, M.; Grupe, K.; Scherneck, S. Estradiol (E2) improves glucose-stimulated insulin secretion and stabilizes GDM progression in a prediabetic mouse model. Int. J. Mol. Sci. 2022, 23, 6693. [Google Scholar] [CrossRef]
- Hay, W.W., Jr. Placental-fetal glucose exchange and fetal glucose metabolism. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 321. [Google Scholar]
- Herrera, E.; Amusquivar, E.; Lopez-Soldado, I.; Ortega, H. Maternal lipid metabolism and placental lipid transfer. Horm. Res. Paediatr. 2006, 65, 59–64. [Google Scholar] [CrossRef]
- Smith, R.; Walsh, A. Composition of liver lipids of the rat during pregnancy and lactation. Lipids 1975, 10, 643–645. [Google Scholar] [CrossRef]
- Jimenez, D.M.; Pocovi, M.; Ramon-Cajal, J.; Romero, M.A.; Martinez, H.; Grande, F. Longitudinal study of plasma lipids and lipoprotein cholesterol in normal pregnancy and puerperium. Gynecol. Obstet. Investig. 1988, 25, 158–164. [Google Scholar] [CrossRef]
- Emet, T.; Üstüner, I.; Güven, S.G.; Balık, G.; Ural, Ü.M.; Tekin, Y.B.; Şentürk, Ş.; Şahin, F.K.; Avşar, A.F. Plasma lipids and lipoproteins during pregnancy and related pregnancy outcomes. Arch. Gynecol. Obstet. 2013, 288, 49–55. [Google Scholar] [CrossRef]
- Sivan, E.; Boden, G. Free fatty acids, insulin resistance, and pregnancy. Curr. Diabetes Rep. 2003, 3, 319–322. [Google Scholar] [CrossRef]
- Villafan-Bernal, J.R.; Acevedo-Alba, M.; Reyes-Pavon, R.; Diaz-Parra, G.A.; Lip-Sosa, D.L.; Vazquez-Delfin, H.I.; Hernandez-Muñoz, M.; Bravo-Aguirre, D.E.; Figueras, F.; Martinez-Portilla, R.J. Plasma levels of free fatty acids in women with gestational diabetes and its intrinsic and extrinsic determinants: Systematic review and meta-analysis. J. Diabetes Res. 2019, 2019, 7098470. [Google Scholar] [CrossRef] [Green Version]
- Boden, G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc. Assoc. Am. Physicians 1999, 111, 241–248. [Google Scholar] [CrossRef]
- Jensen, M.D.; Haymond, M.W.; Rizza, R.A.; Cryer, P.E.; Miles, J. Influence of body fat distribution on free fatty acid metabolism in obesity. J. Clin. Investig. 1989, 83, 1168–1173. [Google Scholar] [CrossRef]
- Liebmann, M.; Grupe, K.; Asuaje Pfeifer, M.; Rustenbeck, I.; Scherneck, S. Differences in lipid metabolism in acquired versus preexisting glucose intolerance during gestation: Role of free fatty acids and sphingosine-1-phosphate. Lipids Health Dis. 2022, 21, 99. [Google Scholar] [CrossRef]
- Puchałowicz, K.; Rać, M.E. The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications-Update in Pathogenesis, Treatment and Monitoring. Cells 2020, 9, 1877. [Google Scholar] [CrossRef]
- Teboul, L.; Febbraio, M.; Gaillard, D.; AMRI, E.-Z.; Silverstein, R.; Grimaldi, P.A. Structural and functional characterization of the mouse fatty acid translocase promoter: Activation during adipose differentiation. Biochem. J. 2001, 360, 305–312. [Google Scholar] [CrossRef]
- Mekada, K.; Abe, K.; Murakami, A.; Nakamura, S.; Nakata, H.; Moriwaki, K.; Obata, Y.; Yoshiki, A. Genetic differences among C57BL/6 substrains. Exp. Anim. 2009, 58, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, D.A.; Davis, D.B. Attention to Background Strain Is Essential for Metabolic Research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes 2016, 65, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, A.; Reifsnyder, P.C.; Malcolm, R.D.; Lucas, C.A.; MacGregor, G.R.; Zhang, W.; Leiter, E.H. Diet-induced obesity in Two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity 2010, 18, 1902–1905. [Google Scholar] [CrossRef] [Green Version]
- Fergusson, G.; Ethier, M.; Guévremont, M.; Chrétien, C.; Attané, C.; Joly, E.; Fioramonti, X.; Prentki, M.; Poitout, V.; Alquier, T. Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Mol. Metab. 2014, 3, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Toye, A.; Lippiat, J.; Proks, P.; Shimomura, K.; Bentley, L.; Hugill, A.; Mijat, V.; Goldsworthy, M.; Moir, L.; Haynes, A. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005, 48, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furse, S.; Fernandez-Twinn, D.S.; Beeson, J.H.; Chiarugi, D.; Ozanne, S.E.; Koulman, A. A mouse model of gestational diabetes shows dysregulated lipid metabolism post-weaning, after return to euglycaemia. Nutr. Diabetes 2022, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- McIlvride, S.; Nikolova, V.; Fan, H.M.; McDonald, J.A.; Wahlström, A.; Bellafante, E.; Jansen, E.; Adorini, L.; Shapiro, D.; Jones, P. Obeticholic acid ameliorates dyslipidemia but not glucose tolerance in mouse model of gestational diabetes. Am. J. Physiol.-Endocrinol. Metab. 2019, 317, E399–E410. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, M.; Rim, J.S.; Koza, R.A.; Kozak, L.P. Variation in type 2 diabetes-related traits in mouse strains susceptible to diet-induced obesity. Diabetes 2003, 52, 1958–1966. [Google Scholar] [CrossRef] [Green Version]
- Andrikopoulos, S.; Blair, A.R.; Deluca, N.; Fam, B.C.; Proietto, J. Evaluating the glucose tolerance test in mice. Am. J. Physiol. -Endocrinol. Metab. 2008, 295, E1323–E1332. [Google Scholar] [CrossRef] [Green Version]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sánchez-Campos, S.; García-Mediavilla, M.V.; Fernández-Bermejo, M.; Lozano-Rodríguez, T.; Vargas-Castrillón, J.; Buqué, X.; Ochoa, B.; Aspichueta, P. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef]
- Koonen, D.P.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.-L.M.; Ong, H.; Vance, D.E.; Dyck, J.R. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar] [CrossRef] [Green Version]
- Jensen, V.S.; Porsgaard, T.; Lykkesfeldt, J.; Hvid, H. Rodent model choice has major impact on variability of standard preclinical readouts associated with diabetes and obesity research. Am. J. Transl. Res. 2016, 8, 3574. [Google Scholar]
- Tuttle, A.H.; Philip, V.M.; Chesler, E.J.; Mogil, J.S. Comparing phenotypic variation between inbred and outbred mice. Nat. Methods 2018, 15, 994–996. [Google Scholar] [CrossRef]
- Taft, R.A.; Davisson, M.; Wiles, M.V. Know thy mouse. Trends Genet. 2006, 22, 649–653. [Google Scholar] [CrossRef]
- Matysková, R.; Maletinska, L.; Maixnerová, J.; Pirnik, Z.; Kiss, A.; Železná, B. Comparison of the obesity phenotypes related to monosodium glutamate effect on arcuate nucleus and/or the high fat diet feeding in C57BL/6 and NMRI mice. Physiol. Res. 2008, 57, 727. [Google Scholar] [CrossRef]
- Nielsen, J.H.; Svensson, C.; Galsgaard, E.D.; Møldrup, A.; Billestrup, N. Beta cell proliferation and growth factors. J. Mol. Med. 1999, 77, 62–66. [Google Scholar] [CrossRef]
- Rieck, S.; Kaestner, K.H. Expansion of β-cell mass in response to pregnancy. Trends Endocrinol. Metab. 2010, 21, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Beamish, C.A.; Zhang, L.; Szlapinski, S.K.; Strutt, B.J.; Hill, D.J. An increase in immature β-cells lacking Glut2 precedes the expansion of β-cell mass in the pregnant mouse. PLoS ONE 2017, 12, e0182256. [Google Scholar] [CrossRef] [Green Version]
- Sorenson, R.L.; Brelje, T.C. Adaptation of islets of Langerhans to pregnancy: Beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm. Metab. Res. 1997, 29, 301–307. [Google Scholar] [CrossRef]
- Genevay, M.; Pontes, H.; Meda, P. Beta cell adaptation in pregnancy: A major difference between humans and rodents? Diabetologia 2010, 53, 2089–2092. [Google Scholar] [CrossRef] [Green Version]
- Green, I.; Taylor, K. Effects of pregnancy in the rat on the size and insulin secretory response of the islets of Langerhans. J. Endocrinol. 1972, 54, 317–325. [Google Scholar] [CrossRef]
- Weinhaus, A.J.; Stout, L.E.; Bhagroo, N.V.; Brelje, T.C.; Sorenson, R.L. Regulation of glucokinase in pancreatic islets by prolactin: A mechanism for increasing glucose-stimulated insulin secretion during pregnancy. J. Endocrinol. 2007, 193, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Sorenson, R.L.; Brelje, T.C.; Hegre, O.D.; Marshall, S.; Anaya, P.; Sheridan, J.D. Prolactin (in vitro) decreases the glucose stimulation threshold, enhances insulin secretion, and increases dye coupling among islet B cells. Endocrinology 1987, 121, 1447–1453. [Google Scholar] [CrossRef]
- Panten, U.; Willenborg, M.; Schumacher, K.; Hamada, A.; Ghaly, H.; Rustenbeck, I. Acute metabolic amplification of insulin secretion in mouse islets is mediated by mitochondrial export of metabolites, but not by mitochondrial energy generation. Metab. Clin. Exp. 2013, 62, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Schulze, T.; Morsi, M.; Reckers, K.; Brüning, D.; Seemann, N.; Panten, U.; Rustenbeck, I. Metabolic amplification of insulin secretion is differentially desensitized by depolarization in the absence of exogenous fuels. Metab. Clin. Exp. 2017, 67, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hatlapatka, K.; Willenborg, M.; Rustenbeck, I. Plasma membrane depolarization as a determinant of the first phase of insulin secretion. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E315–E322. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | Encoded Protein | Protein Function | Reference |
|---|---|---|---|
| CDKAL1 | CDK5 regulatory subunit associated protein 1 like 1 | regulation of β-cell function and glucose-stimulated insulin secretion; associated with impaired insulin secretory capacity | [23,25,26,27,28] |
| CDKN2AB | cyclin-dependent kinase inhibitor 2A/B | regulation of cell proliferation and apoptosis; control of glucose homeostasis, insulin secretion and β-cell function | [9,23,29,30] |
| GCK | glucokinase | phosphorylation of glucose in pancreatic β-cells and hepatocytes; involved in the regulation of insulin secretion; conversion to glycogen in the liver; MODY2 gene | [21,23,25,31] |
| IGF2BP2 | insulin like growth factor 2 mRNA binding protein 2 | modulates cellular metabolism by post transcriptional regulation; associated with impaired β-cell function | [22,23,32] |
| IRS1 | insulin receptor substrate 1 | important role in insulin signaling pathways | [22,31] |
| KCNJ11 | potassium inwardly rectifying channel subfamily J member 11 | encodes the inward-rectifier potassium ion channel (Kir6.2); regulation of insulin secretion | [21,31,33] |
| KCNQ1 | potassium voltage-gated channel subfamily Q member 1 | regulation of insulin secretion; associated with impaired β-cell function | [21,34] |
| MTNR1B | melatonin receptor 1B | regulation of insulin secretion; associated with impaired β-cell function | [21,22,35] |
| TCF7L2 | transcription factor 7 like 2 | transcription factor involved in WNT signaling pathway; associated with IR and impaired insulin secretion | [21,22,36] |
| Model/Strategy | Advantages | Disadvantages |
|---|---|---|
| Surgically induced by pancreatectomy | Suitable when other methods are not an option, especially for larger animals | Not accurately resembling the etiology of the human disease |
| Studying the fetal development affected by changes in the uterine environment | Causes severe and irreversible hyperglycemia, not adequately reflecting the transient glucose intolerance of GDM | |
| Chemically induced using Streptozotocin or Alloxan | Effective, affordable, time saving and widely used method | Not accurately resembling the etiology of the human disease Causes severe and irreversible hyperglycemia, not adequately reflecting the transient glucose intolerance of GDM |
| Nutritional manipulation: administration of a high-fat or high-fat high-sugar diet; glucose infusion | Suitable when other methods are not an option, especially for larger animals Similar to the human disease where obesity and lifestyle are major contributors | Disregards the genetic factors associated with the disease Leads to a condition similar to T2DM with a marked insulin resistance |
| Genetic manipulation | Investigation of β-cell adaptation mechanisms during gestation Studying the genetic mechanisms involved in human disease | Limited to specific animal models Large number of animals used for the generation of knockout models Inadequately reflects the complex interaction between polygenic and environmental factors |
| Polygenic models: e.g., New Zealand obese (NZO) mouse | Reflects the polygenic character of the disease Suitable for the characterization of a subpopulation of human GDM exhibiting IGT prior to pregnancy or prediabetes | Preconceptional IGT not consistent with the conventional GDM definition |
| Acquired model: C57BL/6N | Shows IGT during gestation in the absence of an intervention | Controversial model which is commonly used as a control strain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grupe, K.; Scherneck, S. Mouse Models of Gestational Diabetes Mellitus and Its Subtypes: Recent Insights and Pitfalls. Int. J. Mol. Sci. 2023, 24, 5982. https://doi.org/10.3390/ijms24065982
Grupe K, Scherneck S. Mouse Models of Gestational Diabetes Mellitus and Its Subtypes: Recent Insights and Pitfalls. International Journal of Molecular Sciences. 2023; 24(6):5982. https://doi.org/10.3390/ijms24065982
Chicago/Turabian StyleGrupe, Katharina, and Stephan Scherneck. 2023. "Mouse Models of Gestational Diabetes Mellitus and Its Subtypes: Recent Insights and Pitfalls" International Journal of Molecular Sciences 24, no. 6: 5982. https://doi.org/10.3390/ijms24065982
APA StyleGrupe, K., & Scherneck, S. (2023). Mouse Models of Gestational Diabetes Mellitus and Its Subtypes: Recent Insights and Pitfalls. International Journal of Molecular Sciences, 24(6), 5982. https://doi.org/10.3390/ijms24065982