
Mouse Models with SGLT2 Mutations: Toward Understanding the Role of SGLT2 beyond Glucose Reabsorption

 , and
, and 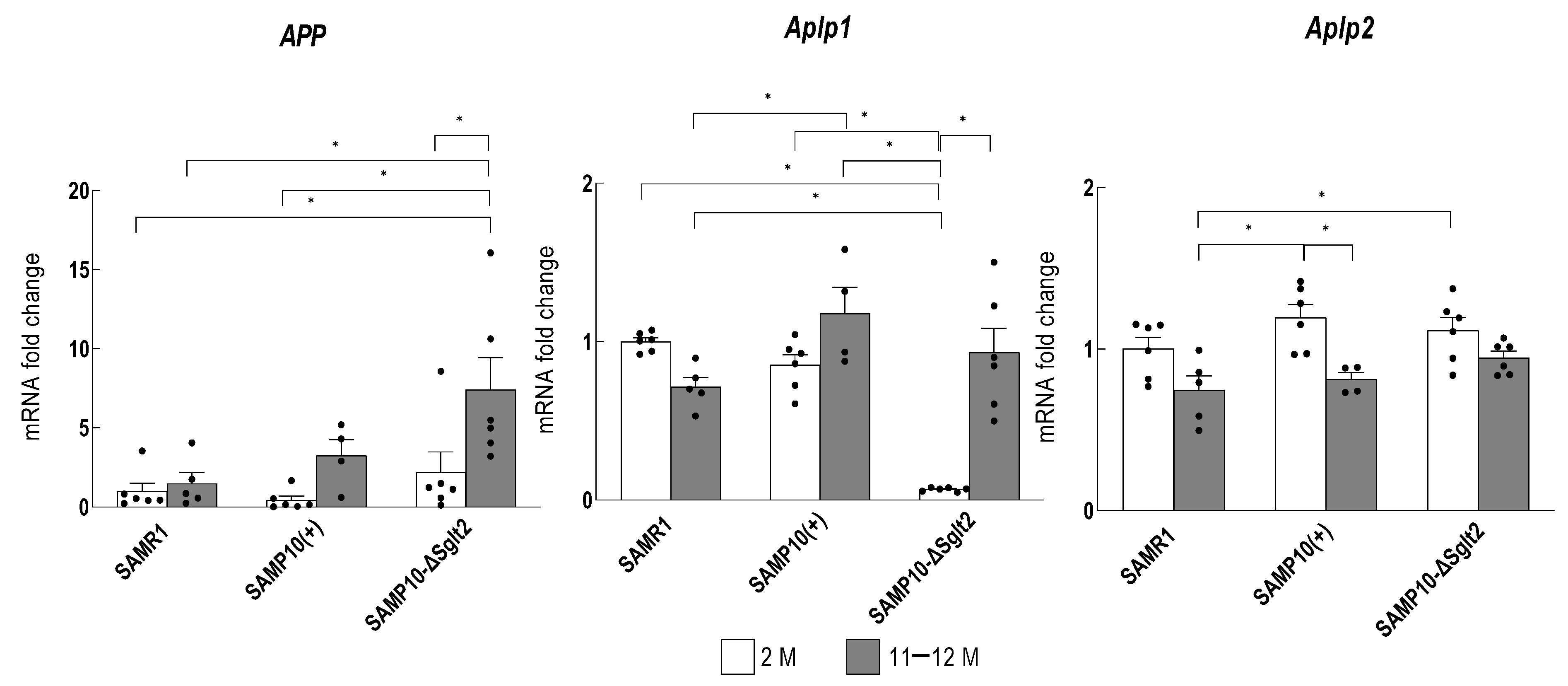{kind=link}
Abstract
:1. Introduction
2. Renal Glucosuria in Humans
2.1. SGLT2 Mutation and SGLT2 Inhibitor
2.2. Other Kidney Glucose Transporters and Their Diseases
3. Renal Glucosuria Models in Experimental Animals
3.1. SGLT2−/− Mouse
3.2. Sweet Pee Mouse
3.3. Jimbee Mouse
3.4. SAMP10-ΔSglt2 Mouse
Amyloid Precursor Protein (APP) and Amyloid Precursor-like Protein (Aplp) in the Hippocampus of SAMP10-ΔSglr2
4. Discussion
5. Conclusions
- SGLT2 mutant mice are valuable as a means of demonstrating the efficacy of SGLT2 inhibition.
- SGLT mutation and inhibition may increase the risk of malfunction.
- The mutant mice are important for information on changes in bone and mineral metabolism due to SGLT2 inhibition.
- The mutant mouse is an interesting model for investigating the relationship between glucose metabolism in the brain and Alzheimer’s disease.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Gylfe, E.; Gilon, P. Glucose regulation of glucagon secretion. Diabetes Res. Clin. Pract. 2014, 103, 1–10. [Google Scholar] [CrossRef]
- Mitrakou, A. Kidney: Its impact on glucose homeostasis and hormonal regulation. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. S1), S66–S72. [Google Scholar] [CrossRef] [PubMed]
- Sędzikowska, A.; Szablewski, L. Human Glucose Transporters in Renal Glucose Homeostasis. Int. J. Mol. Sci. 2021, 22, 13522. [Google Scholar] [CrossRef] [PubMed]
- Wallner, E.I.; Wada, J.; Tramonti, G.; Lin, S.; Kanwar, Y.S. Status of glucose transporters in the mammalian kidney and renal development. Ren. Fail. 2001, 23, 301–310. [Google Scholar] [CrossRef]
- Shepard, B.D.; Pluznick, J.L. Saving the sweetness: Renal glucose handling in health and disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F55–F61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aires, I.; Fila, M.; Polidori, D.; Santos, A.R.; Costa, A.B.; Calado, J. Determination of the renal threshold for glucose excretion in Familial Renal Glucosuria. Nephron 2015, 129, 300–304. [Google Scholar] [CrossRef]
- Kleta, R.; Stuart, C.; Gill, F.A.; Gahl, W.A. Renal glucosuria due to SGLT2 mutations. Mol. Genet. Metab. 2004, 82, 56–58. [Google Scholar] [CrossRef]
- Calado, J.; Loeffler, J.; Sakallioglu, O.; Gok, F.; Lhotta, K.; Barata, J.; Rueff, J. Familial renal glucosuria: SLC5A2 mutation analysis and evidence of salt-wasting. Kidney Int. 2006, 69, 852–855. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, Y.; Huang, L.; Kong, M.; Yang, Z. A novel compound heterozygous mutation in SLC5A2 contributes to familial renal glucosuria in a Chinese family, and a review of the relevant literature. Mol. Med. Rep. 2019, 19, 4364–4376. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, M.; Wang, T.; Zhang, H.; Ping, F.; Zhang, Q.; Xu, J.; Feng, K.; Xiao, X. Genetic analysis and literature review of Chinese patients with familial renal glucosuria: Identification of a novel SLC5A2 mutation. Clin. Chim. Acta 2017, 469, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Santer, R.; Calado, J. Familial renal glucosuria and SGLT2: From a mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. 2010, 5, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishman, B.; Shlomai, G.; Twig, G.; Derazne, E.; Tenenbaum, A.; Fisman, E.Z.; Leiba, A.; Grossman, E. Renal glucosuria is associated with lower body weight and lower rates of elevated systolic blood pressure: Results of a nationwide cross-sectional study of 2.5 million adolescents. Cardiovasc. Diabetol. 2019, 18, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Han, K.H.; Park, H.W.; Shin, J.I.; Kim, C.J.; Namgung, M.K.; Kim, K.H.; Koo, J.W.; Chung, W.Y.; Lee, D.Y.; et al. Familial renal glucosuria: A clinicogenetic study of 23 additional cases. Pediatr. Nephrol. 2012, 27, 1091–1095. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, X.; Zhang, R.; Wang, C.; Han, Y.; Shao, L. Identification of ten novel SLC5A2 mutations and determination of the renal threshold for glucose excretion in Chinese patients with familial renal glucosuria. Clin. Chim. Acta 2019, 490, 102–106. [Google Scholar] [CrossRef]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Ly, J.P.; Onay, T.; Sison, K.; Sivaskandarajah, G.; Sabbisetti, V.; Li, L.; Bonventre, J.V.; Flenniken, A.; Paragas, N.; Barasch, J.M.; et al. The Sweet Pee model for Sglt2 mutation. J. Am. Soc. Nephrol. 2011, 22, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Jurczak, M.J.; Lee, H.Y.; Birkenfeld, A.L.; Jornayvaz, F.R.; Frederick, D.W.; Pongratz, R.L.; Zhao, X.; Moeckel, G.W.; Samuel, V.T.; Whaley, J.M.; et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes 2011, 60, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Thrailkill, K.M.; Bunn, R.C.; Uppuganti, S.; Ray, P.; Garrett, K.; Popescu, I.; Pennings, J.S.; Fowlkes, J.L.; Nyman, J.S. Genetic ablation of SGLT2 function in mice impairs tissue mineral density but does not affect fracture resistance of bone. Bone 2020, 133, 115254. [Google Scholar] [CrossRef]
- Unno, K.; Yamamoto, H.; Toda, M.; Hagiwara, S.; Iguchi, K.; Hoshino, M.; Takabayashi, F.; Hasegawa-Ishii, S.; Shimada, A.; Hosokawa, M.; et al. Novel frame-shift mutation in Slc5a2 encoding SGLT2 in a strain of senescence-accelerated mouse SAMP10. Biochem. Biophys. Res. Commun. 2014, 454, 89–94. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Cole, J.B.; Florez, J.C. Genetics of diabetes mellitus and diabetes complications. Nat. Rev. Nephrol. 2020, 6, 377–390. [Google Scholar] [CrossRef]
- Garcia-Ropero, A.; Badimon, J.J.; Santos-Gallego, C.G. The pharmacokinetics and pharmacodynamics of SGLT2 inhibitors for type 2 diabetes mellitus: The latest developments. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Rieg, T.; Vallon, V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia 2018, 61, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, Y.; Matsui, T.; Yamagishi, S.; Tofogliflozin, A. Highly Selective Inhibitor of SGLT2 Blocks Proinflammatory and Proapoptotic Effects of Glucose Overload on Proximal Tubular Cells Partly by Suppressing Oxidative Stress Generation. Horm. Metab. Res. 2016, 48, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Van der Aart-van der Beek, A.B.; de Boer, R.A.; Heerspink, H.J.L. Kidney and heart failure outcomes associated with SGLT2 inhibitor use. Nat. Rev. Nephrol. 2022, 18, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sridhar, V.S.; Boulet, J.; Dharia, A.; Khan, A.; Lawler, P.R.; Cherney, D.Z.I. Cardiorenal protection with SGLT2 inhibitors in patients with diabetes mellitus: From biomarkers to clinical outcomes in heart failure and diabetic kidney disease. Metabolism 2022, 126, 154918. [Google Scholar] [CrossRef]
- Hou, Y.C.; Zheng, C.M.; Yen, T.H.; Lu, K.C. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. Int. J. Mol. Sci. 2020, 21, 7833. [Google Scholar] [CrossRef]
- Sha, W.; Wen, S.; Chen, L.; Xu, B.; Lei, T.; Zhou, L. The Role of SGLT2 Inhibitor on the Treatment of Diabetic Retinopathy. J. Diabetes Res. 2020, 2020, 8867875. [Google Scholar] [CrossRef]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434, Erratum in J. Am. Coll. Cardiol. 2020, 76, 1505. [Google Scholar] [CrossRef]
- Fathi, A.; Vickneson, K.; Singh, J.S. SGLT2-inhibitors; More than just glycosuria and diuresis. Heart Fail. Rev. 2021, 26, 623–642. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadi, A.; Bellia, A.; Di Daniele, N.; Meloni, M.; Lauro, R.; Della-Morte, D.; Lauro, D. The molecular link between oxidative stress, insulin resistance, and type 2 diabetes: A target for new therapies against cardiovascular diseases. Curr. Opin. Pharmacol. 2022, 62, 85–96. [Google Scholar] [CrossRef]
- Goldenberg, R.M.; Berard, L.D.; Cheng, A.Y.Y.; Gilbert, J.D.; Verma, S.; Woo, V.C.; Yale, J.F. SGLT2 Inhibitor-associated Diabetic Ketoacidosis: Clinical Review and Recommendations for Prevention and Diagnosis. Clin. Ther. 2016, 38, 2654–2664.e1. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, A. Risks Associated with SGLT2 Inhibitors: An Overview. Curr. Drug Saf. 2018, 13, 84–91. [Google Scholar] [CrossRef]
- Ottosson-Laakso, E.; Tuomi, T.; Forsén, B.; Gullström, M.; Groop, P.H.; Groop, L.; Vikman, P. Influence of Familial Renal Glycosuria Due to Mutations in the SLC5A2 Gene on Changes in Glucose Tolerance over Time. PLoS ONE 2016, 11, e0146114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.L.; Perazella, M.A. SGLT2 inhibitor therapy in patients with type-2 diabetes mellitus: Is acute kidney injury a concern? J. Nephrol. 2020, 33, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.; Yousuf, S.; Singh, M.P. Contributive Role of Hyperglycemia and Hypoglycemia Towards the Development of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 4274–4291. [Google Scholar] [CrossRef]
- Needham, B.E.; Wlodek, M.E.; Ciccotosto, G.D.; Fam, B.C.; Masters, C.L.; Proietto, J.; Andrikopoulos, S.; Cappai, R. Identification of the Alzheimer’s disease amyloid precursor protein (APP) and its homologue APLP2 as essential modulators of glucose and insulin homeostasis and growth. J. Pathol. 2008, 215, 155–163. [Google Scholar] [CrossRef]
- Tu, Z.; Keller, M.P.; Zhang, C.; Rabaglia, M.E.; Greenawalt, D.M.; Yang, X.; Wang, I.M.; Dai, H.; Bruss, M.D.; Lum, P.Y.; et al. Integrative analysis of a cross-loci regulation network identifies App as a gene regulating insulin secretion from pancreatic islets. PLoS Genet. 2012, 8, e1003107. [Google Scholar] [CrossRef]
- Botteri, G.; Salvadó, L.; Gumà, A.; Hamilton, D.L.; Meakin, P.J.; Montagut, G.; Ashford, M.L.J.; Ceperuelo-Mallafré, V.; Fernández-Veledo, S.; Vendrell, J.; et al. The BACE1 product sAPPβ induces ER stress and inflammation and impairs insulin signaling. Metabolism 2018, 85, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, R.D.; Ou, Y.; Davis, J.E.; Odle, A.K.; Groves, T.R.; Allen, A.R.; Childs, G.V.; Barger, S.W. Alzheimer amyloid-β- peptide disrupts membrane localization of glucose transporter 1 in astrocytes: Implications for glucose levels in brain and blood. Neurobiol. Aging 2021, 97, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Faselis, C.; Katsimardou, A.; Imprialos, K.; Deligkaris, P.; Kallistratos, M.; Dimitriadis, K. Microvascular Complications of Type 2 Diabetes Mellitus. Curr. Vasc. Pharmacol. 2020, 18, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A.; Norton, L. Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30–50% of filtered glucose load in humans. Diabetes 2013, 62, 3324–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Perry, R.J.; Shulman, G.I. Sodium-glucose cotransporter-2 inhibitors: Understanding the mechanisms for therapeutic promise and persisting risks. J. Biol. Chem. 2020, 295, 14379–14390. [Google Scholar] [CrossRef]
- Feijóo-Bandín, S.; Aragón-Herrera, A.; Otero-Santiago, M.; Anido-Varela, L.; Moraña-Fernández, S.; Tarazón, E.; Roselló-Lletí, E.; Portolés, M.; Gualillo, O.; González-Juanatey, J.R.; et al. Role of Sodium-Glucose Co-Transporter 2 Inhibitors in the Regulation of Inflammatory Processes in Animal Models. Int. J. Mol. Sci. 2022, 23, 5634. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiciński, M.; Wódkiewicz, E.; Górski, K.; Walczak, M.; Malinowski, B. Perspective of SGLT2 Inhibition in Treatment of Conditions Connected to Neuronal Loss: Focus on Alzheimer’s Disease and Ischemia-Related Brain Injury. Pharmaceuticals 2020, 13, 379. [Google Scholar] [CrossRef]
- Wright, E.M.; Turk, E.; Martin, M.G. Molecular basis for glucose-galactose malabsorption. Cell. Biochem. Biophys. 2002, 36, 115–121. [Google Scholar] [CrossRef]
- Sharari, S.; Abou-Alloul, M.; Hussain, K.; Ahmad Khan, F. Fanconi-Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia. Int. J. Mol. Sci. 2020, 21, 6286. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Ren. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [Green Version]
- Gerber, C.; Wang, X.; David, V.; Quaggin, S.E.; Isakova, T.; Martin, A. Long-Term Effects of Sglt2 Deletion on Bone and Mineral Metabolism in Mice. JBMR Plus 2021, 5, e10526. [Google Scholar] [CrossRef]
- Hughes, C.B.; Mussman, G.M.; Ray, P.; Bunn, R.C.; Cornea, V.; Thrailkill, K.M.; Fowlkes, J.L.; Popescu, I. Impact of an SGLT2-loss of function mutation on renal architecture, histology, and glucose homeostasis. Cell Tissue Res. 2021, 384, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM). In The Senescence-Accelerated Mouse (SAM): Achievement and Future Directions; Takeda, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 3–14. [Google Scholar]
- Takeda, T.; Hosokawa, M.; Takeshita, S.; Irino, M.; Higuchi, K.; Matsushita, T.; Tomita, Y.; Yasuhira, K.; Hamamoto, H.; Shimizu, K.; et al. A new murine model of accelerated senescence. Mech. Ageing Dev. 1981, 17, 183–194. [Google Scholar] [CrossRef]
- Takeda, T. Senescence-accelerated mouse (SAM) with special references to neurodegeneration models, SAMP8 and SAMP10 mice. Neurochem. Res. 2009, 34, 639–659. [Google Scholar] [CrossRef]
- Unno, K.; Takagi, Y.; Konishi, T.; Suzuki, M.; Miyake, A.; Kurotaki, T.; Hase, T.; Meguro, S.; Shimada, A.; Hasegawa-Ishii, S.; et al. Mutation in Sodium-Glucose Cotransporter 2 Results in Down-Regulation of Amyloid Beta (A4) Precursor-Like Protein 1 in Young Age, Which May Lead to Poor Memory Retention in Old Age. Int. J. Mol. Sci. 2020, 21, 5579. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhu, L.; Tan, J.; Chen, K.; Yu, B. Suppression of miR-130a-3p Attenuates Oxygen-Glucose Deprivation/Reoxygenation-Induced Dendritic Spine Loss by Promoting APP. Front. Neurosci. 2021, 15, 601850. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, C.; Colangelo, L.; Santori, R.; Renella, M.; Mastrantonio, M.; Minisola, S.; Pepe, J. The Interplay Between Bone and Glucose Metabolism. Front. Endocrinol. 2020, 11, 122. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Lv, R.; Wang, J.; Che, L.; Wang, Z.; Huai, Z.; Wang, Y.; Xu, L. The Extraglycemic Effect of SGLT-2 is on Mineral and Bone Metabolism and Bone Fracture. Front. Endocrinol. 2022, 13, 918350. [Google Scholar] [CrossRef] [PubMed]
- Vinke, J.S.J.; Heerspink, H.J.L.; de Borst, M.H. Effects of sodium glucose cotransporter 2 inhibitors on mineral metabolism in type 2 diabetes mellitus. Curr. Opin. Nephrol. Hypertens. 2019, 28, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Papachristoforou, E.; Lambadiari, V.; Maratou, E.; Makrilakis, K. Association of Glycemic Indices (Hyperglycemia, Glucose Variability, and Hypoglycemia) with Oxidative Stress and Diabetic Complications. J. Diabetes Res. 2020, 2020, 7489795. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.E.; Gilsanz, P.; Eng, C.; Beeri, M.S.; Karter, A.J.; Whitmer, R.A. Severe Hypoglycemia and Cognitive Function in Older Adults With Type 1 Diabetes: The Study of Longevity in Diabetes (SOLID). Diabetes Care 2020, 43, 541–548. [Google Scholar] [CrossRef]
- Nevo-Shenker, M.; Shalitin, S. The Impact of Hypo- and Hyperglycemia on Cognition and Brain Development in Young Children with Type 1 Diabetes. Horm. Res. Paediatr. 2021, 94, 115–123. [Google Scholar] [CrossRef]
- Languren, G.; Montiel, T.; Julio-Amilpas, A.; Massieu, L. Neuronal damage and cognitive impairment associated with hypoglycemia: An integrated view. Neurochem. Int. 2013, 63, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Heber, S.; Herms, J.; Gajic, V.; Hainfellner, J.; Aguzzi, A.; Rülicke, T.; von Kretzschmar, H.; von Koch, C.; Sisodia, S.; Tremml, P.; et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 2000, 20, 7951–7963. [Google Scholar] [CrossRef] [Green Version]
- Schilling, S.; Mehr, A.; Ludewig, S.; Stephan, J.; Zimmermann, M.; August, A.; Strecker, P.; Korte, M.; Koo, E.H.; Müller, U.C.; et al. APLP1 Is a Synaptic Cell Adhesion Molecule, Supporting Maintenance of Dendritic Spines and Basal Synaptic Transmission. J. Neurosci. 2017, 37, 5345–5365. [Google Scholar] [CrossRef] [Green Version]
- Onodera, W.; Asahi, T.; Sawamura, N. Rapid evolution of mammalian APLP1 as a synaptic adhesion molecule. Sci. Rep. 2021, 11, 11305. [Google Scholar] [CrossRef]
- Vnencak, M.; Paul, M.H.; Hick, M.; Schwarzacher, S.W.; Del Turco, D.; Müller, U.C.; Deller, T.; Jedlicka, P. Deletion of the amyloid precursor-like protein 1 (APLP1) enhances excitatory synaptic transmission, reduces network inhibition but does not impair synaptic plasticity in the mouse dentate gyrus. J. Comp. Neurol. 2015, 523, 1717–1729. [Google Scholar] [CrossRef]
- Lee, S.H.; Kang, J.; Ho, A.; Watanabe, H.; Bolshakov, V.Y.; Shen, J. APP Family Regulates Neuronal Excitability and Synaptic Plasticity but Not Neuronal Survival. Neuron 2020, 108, 676–690.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Z.; Sun, L.; Yang, L.; Li, H.; Cole, A.L.; Rodriguez-Rivera, J.; Lu, H.C.; Zheng, H. The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J. Neurosci. 2014, 34, 13314–13325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.J.; Yang, M.S.; Zhang, B.; Niu, F.; Dong, J.Q.; Liu, B.Y. Glucose metabolism: A link between traumatic brain injury and Alzheimer’s disease. Chin. J. Traumatol. 2021, 24, 5–10. [Google Scholar] [CrossRef]
- Tomobe, K.; Nomura, Y. Neurochemistry, neuropathology, and heredity in SAMP8: A mouse model of senescence. Neurochem. Res. 2009, 34, 660–669. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Kumar, V.B.; Armbrecht, H.J. The SAMP8 mouse: A model to develop therapeutic interventions for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 1123–1130. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Shi, J.S. SAMP8 Mice as a Model of Age-Related Cognition Decline with Underlying Mechanisms in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 75, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Nishikawa, H.; Hirai, K.; Kato, K.; Miyamoto, M. Relationship of impaired brain glucose metabolism to learning deficit in the senescence-accelerated mouse. Neurosci. Lett. 1996, 217, 37–40. [Google Scholar] [CrossRef]
- Farr, S.A.; Roesler, E.; Niehoff, M.L.; Roby, D.A.; McKee, A.; Morley, J.E. Metformin Improves Learning and Memory in the SAMP8 Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1699–1710. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unno, K.; Taguchi, K.; Takagi, Y.; Hase, T.; Meguro, S.; Nakamura, Y. Mouse Models with SGLT2 Mutations: Toward Understanding the Role of SGLT2 beyond Glucose Reabsorption. Int. J. Mol. Sci. 2023, 24, 6278. https://doi.org/10.3390/ijms24076278
Unno K, Taguchi K, Takagi Y, Hase T, Meguro S, Nakamura Y. Mouse Models with SGLT2 Mutations: Toward Understanding the Role of SGLT2 beyond Glucose Reabsorption. International Journal of Molecular Sciences. 2023; 24(7):6278. https://doi.org/10.3390/ijms24076278
Chicago/Turabian StyleUnno, Keiko, Kyoko Taguchi, Yoshiichi Takagi, Tadashi Hase, Shinichi Meguro, and Yoriyuki Nakamura. 2023. "Mouse Models with SGLT2 Mutations: Toward Understanding the Role of SGLT2 beyond Glucose Reabsorption" International Journal of Molecular Sciences 24, no. 7: 6278. https://doi.org/10.3390/ijms24076278
APA StyleUnno, K., Taguchi, K., Takagi, Y., Hase, T., Meguro, S., & Nakamura, Y. (2023). Mouse Models with SGLT2 Mutations: Toward Understanding the Role of SGLT2 beyond Glucose Reabsorption. International Journal of Molecular Sciences, 24(7), 6278. https://doi.org/10.3390/ijms24076278