

Plumbagin Exhibits Genotoxicity and Induces G2/M Cell Cycle Arrest via ROS-Mediated Oxidative Stress and Activation of ATM-p53 Signaling Pathway in Hepatocellular Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
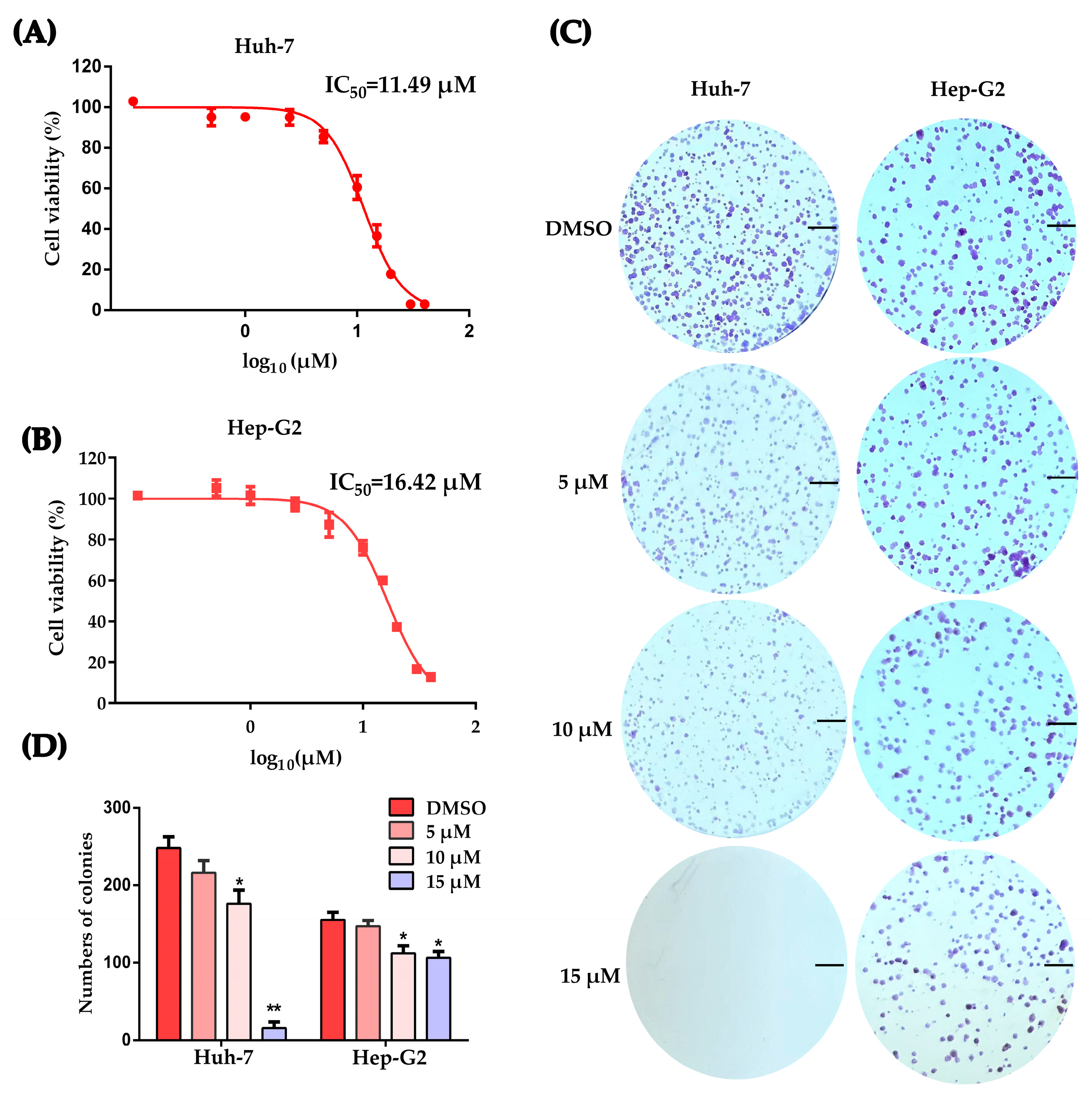
2.1. Working Concentration Determination of PLB on HCC Cells
2.2. Plumbagin Induces G2/M Cell Cycle Arrest of HCC Cells
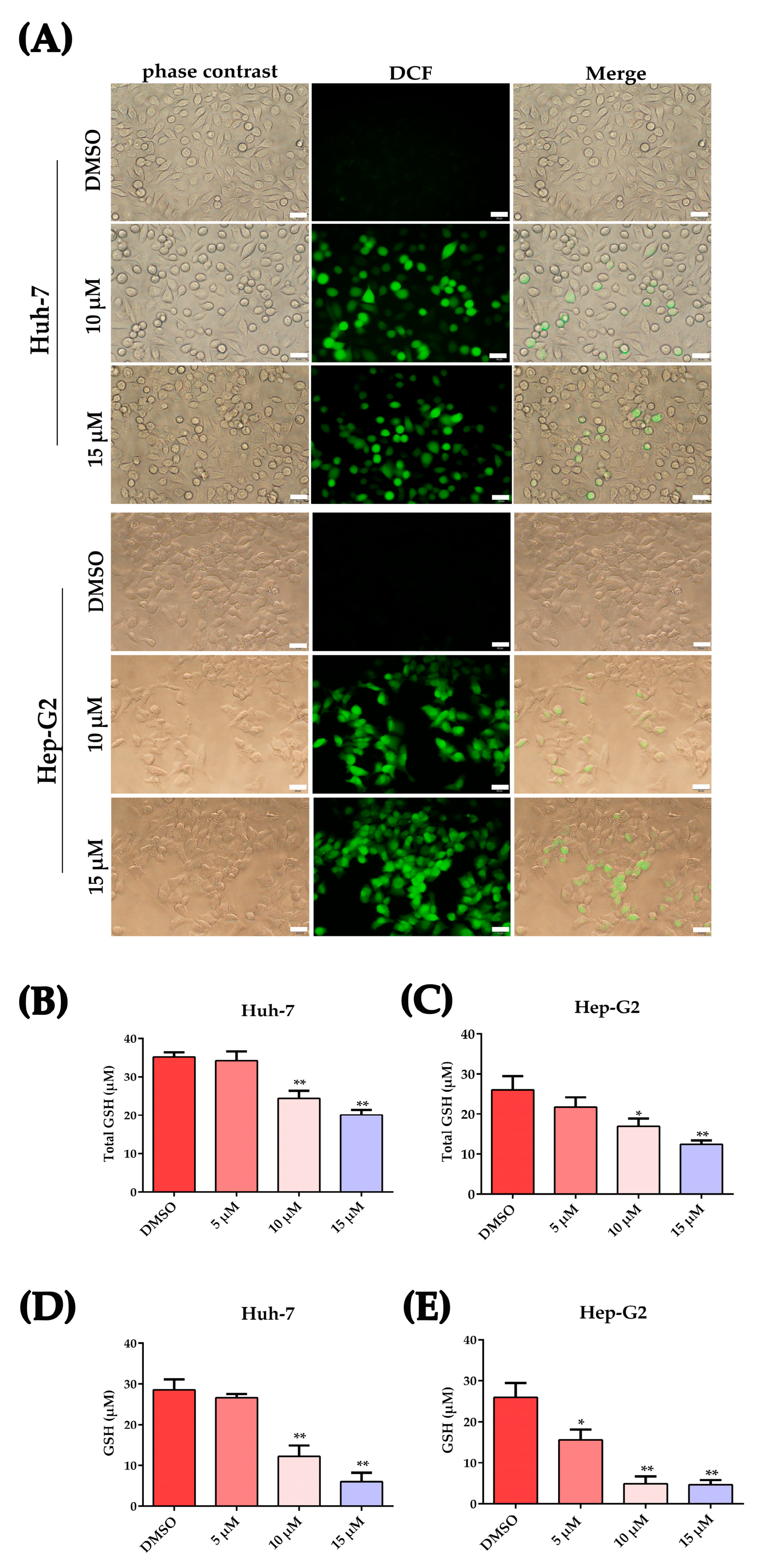
2.3. Plumbagin Increased Oxidative Stress in HCC Cells
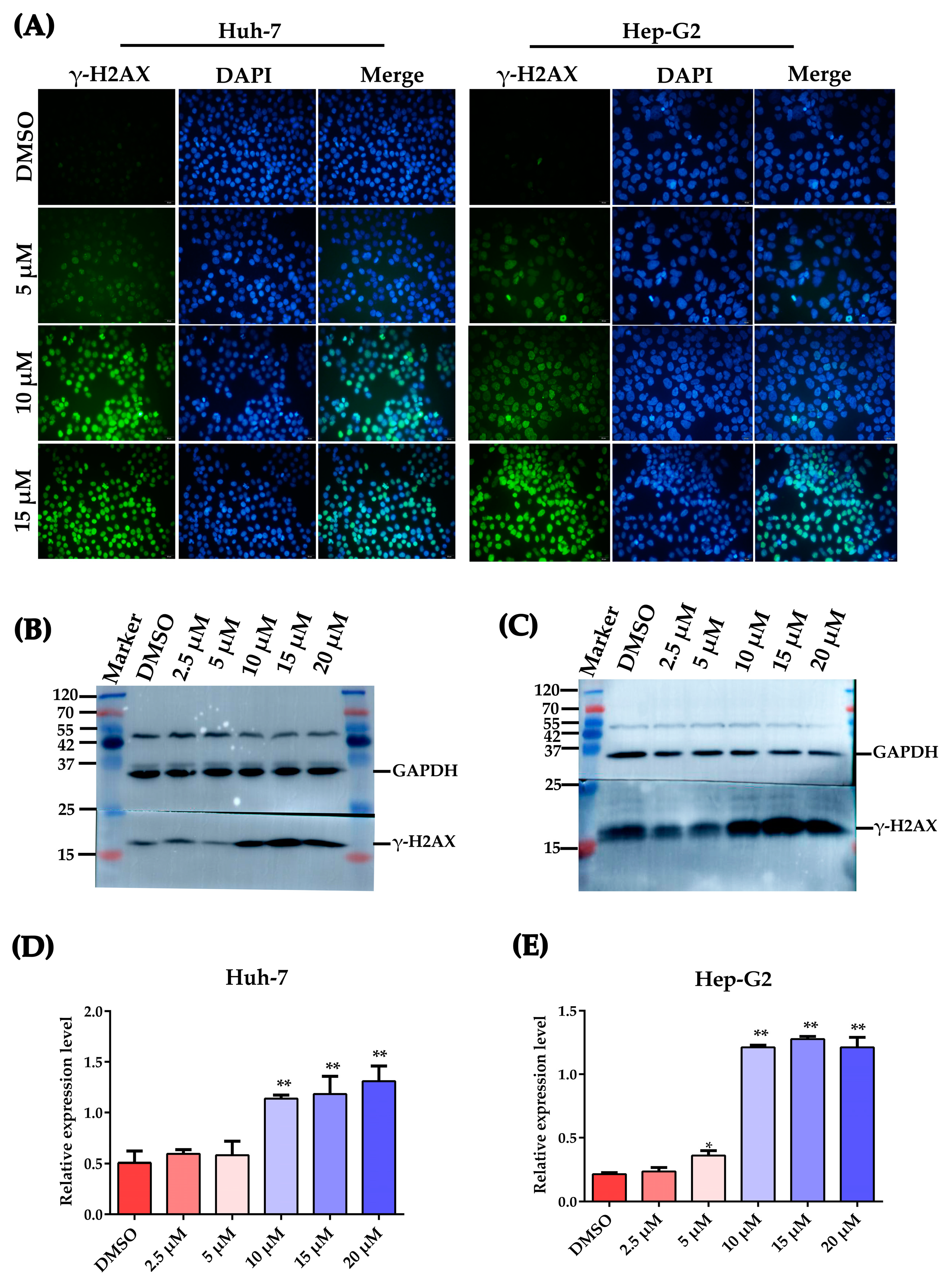
2.4. Plumbagin Induces DNA Damage in HCC Cells
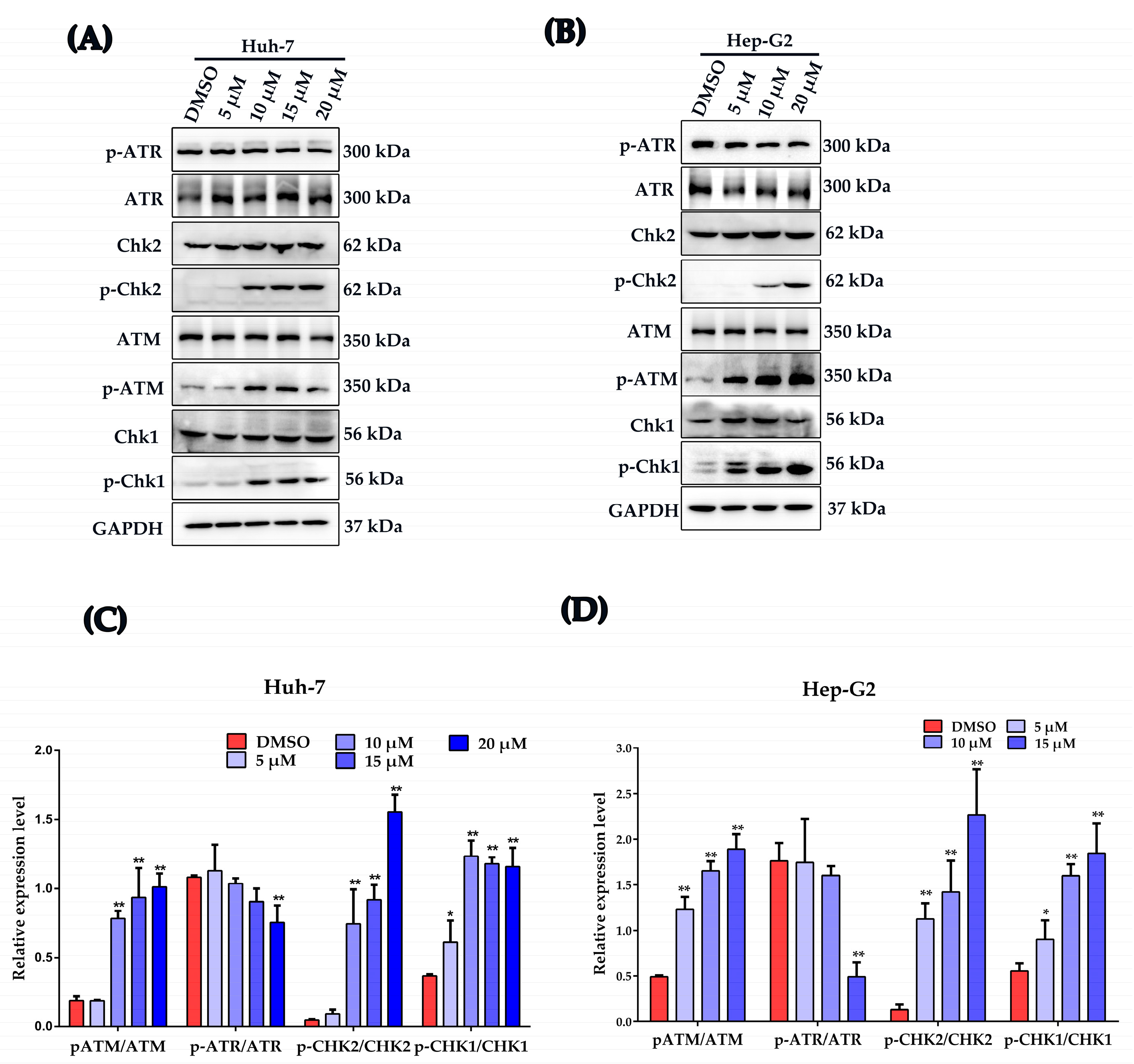
2.5. Plumbagin Activates DNA Damage Response in HCC Cells
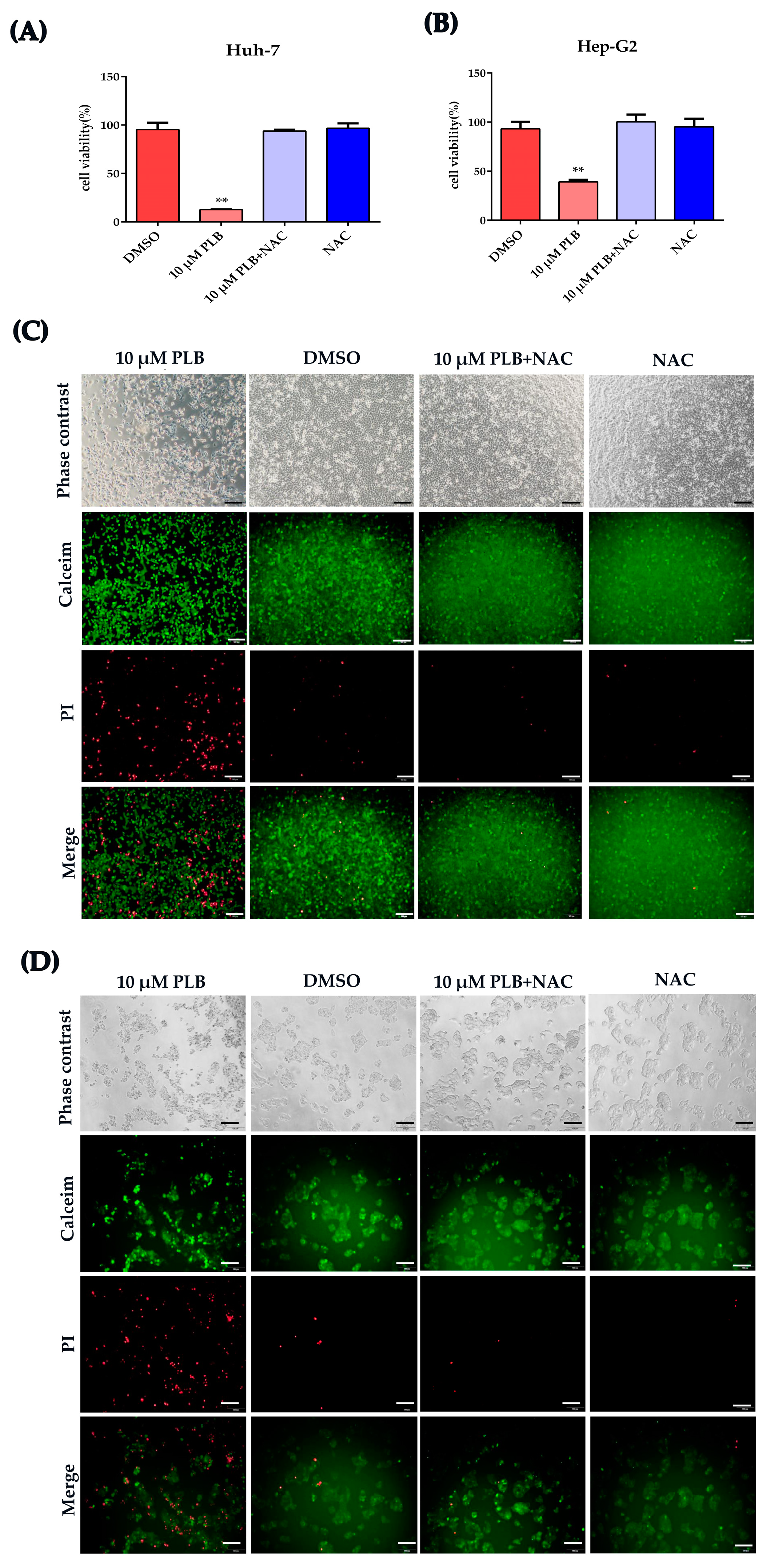
2.6. NAC Treatment Inhibits the Cytotoxicity and DNA Damage Induced by Plumbagin
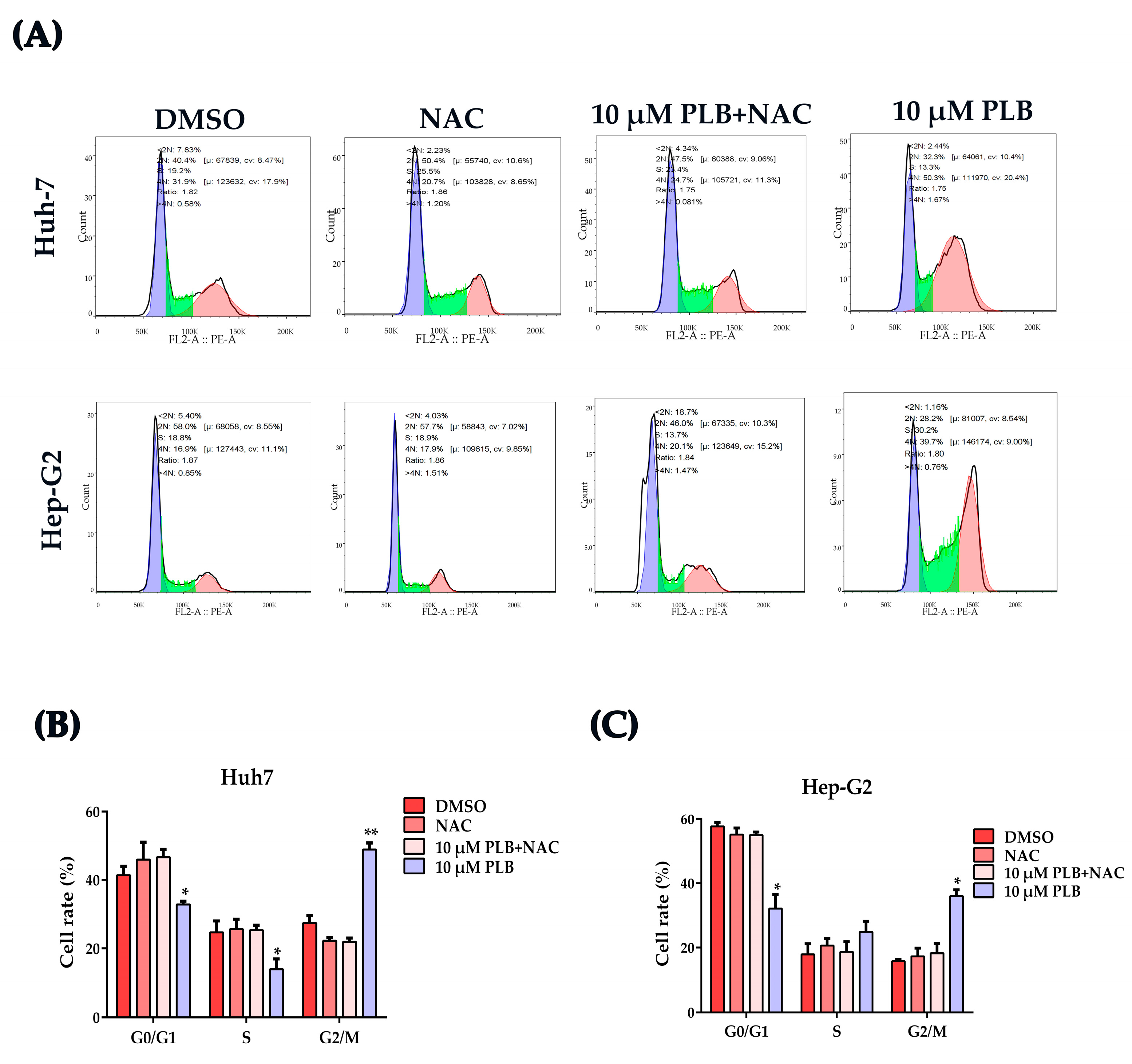
2.7. NAC Treatment Reverses the Cell Cycle Arrest Effects Induced by Plumbagin
2.8. NAC Treatment Inhibits the Activation of ATM-Chk1/2-p53 Signaling Pathway
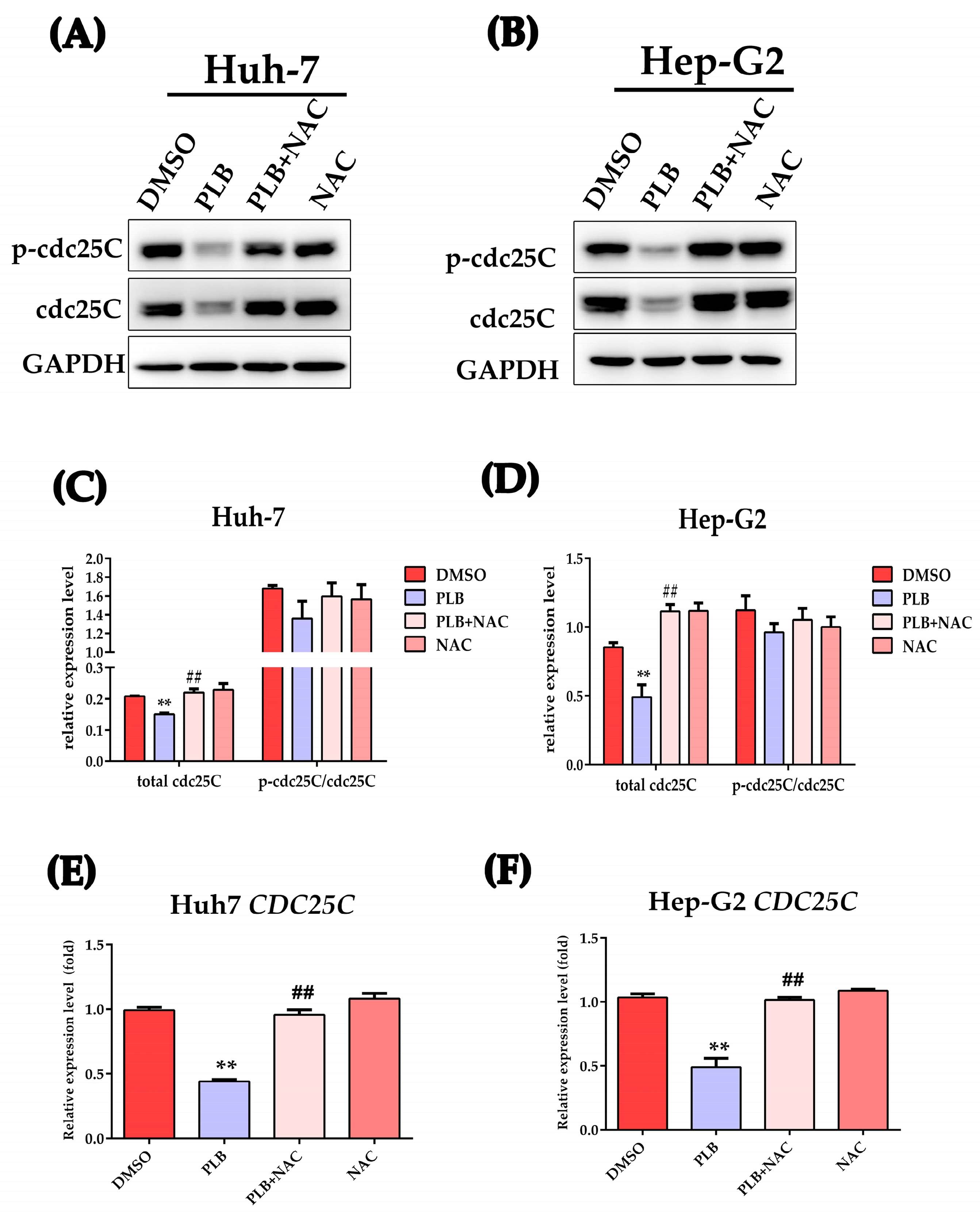
2.9. PLB-Induced ROS-Dependent Downregulation of cdc25C
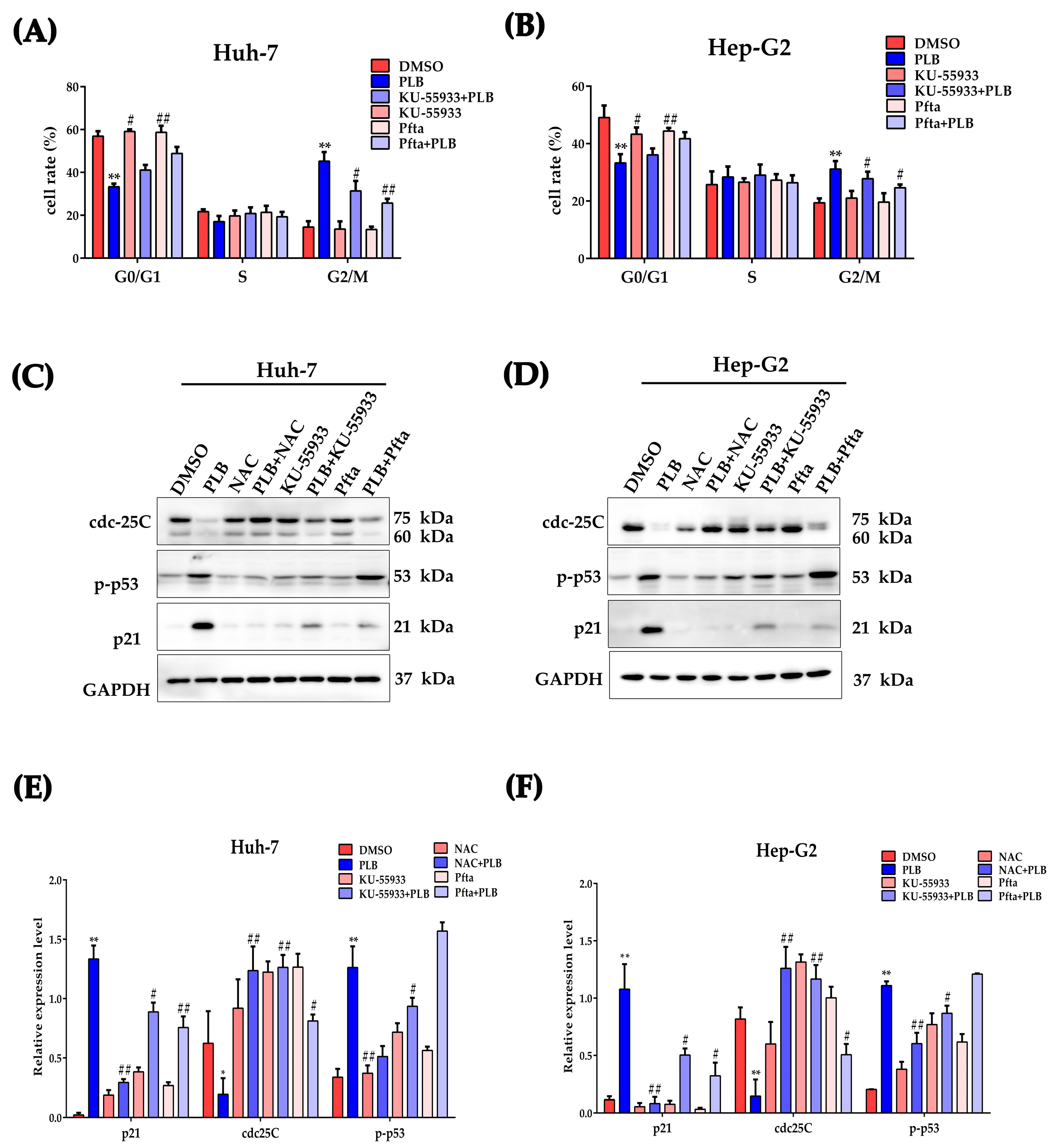
2.10. ATM-p53 Pathway Plays an Important Role in Plumbagin-Induced G2/M Cell Cycle Arrest
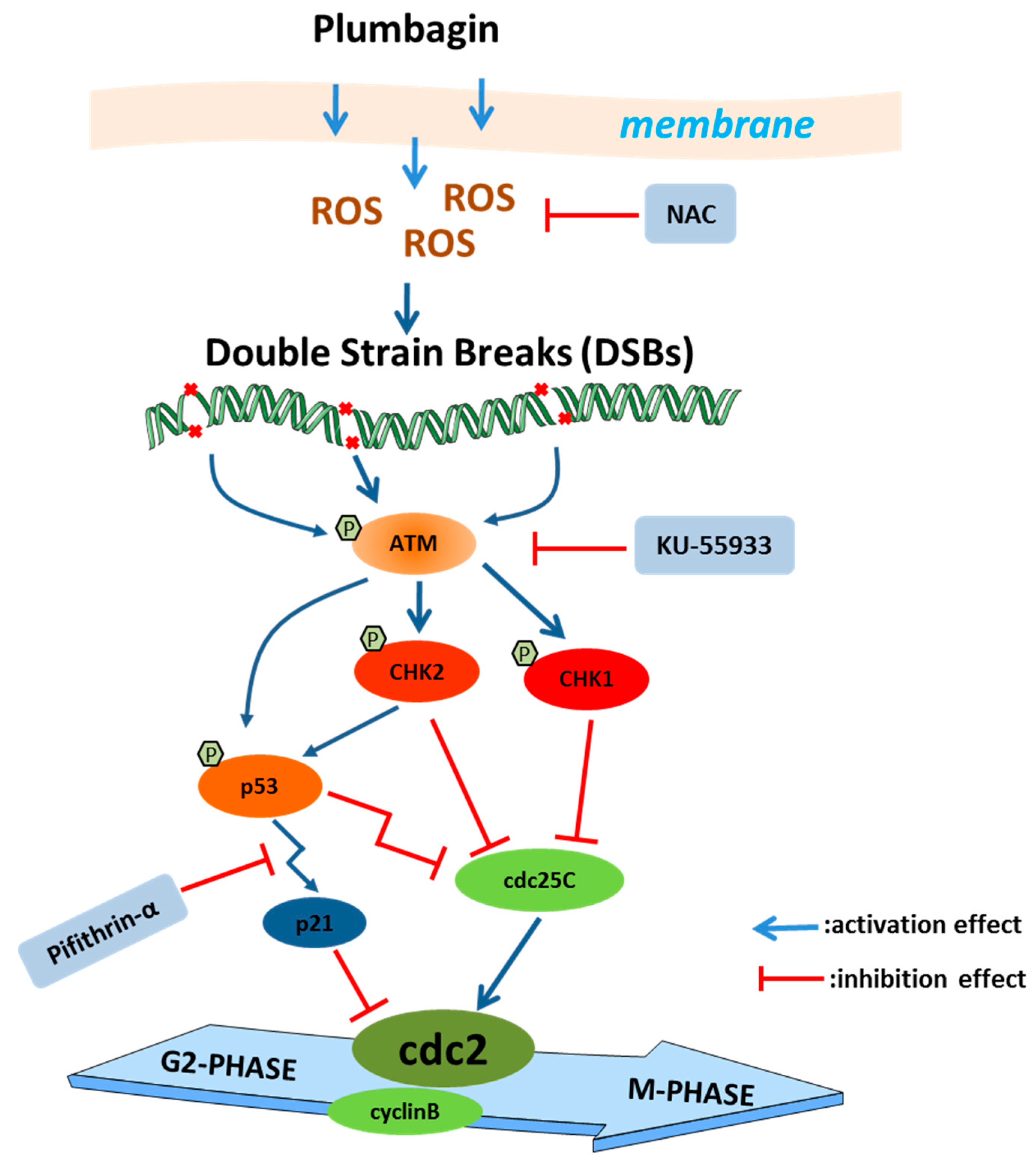
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Chemical Agent
4.2. Cell Viability Assay
4.3. Colony Formation Assay
4.4. Cell Cycle Analysis
4.5. DNA Damage Assay
4.6. ROS Assay
4.7. Measurement of GSH and GSSG
4.8. Western Blotting
4.9. RT-qPCR Method
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rajendran, L.; Ivanics, T.; Claasen, M.P.; Muaddi, H.; Sapisochin, G. The management of post-transplantation recurrence of hepatocellular carcinoma. Clin. Mol. Hepatol. 2022, 28, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, S.; Wu, X.; Zhang, J.; Chen, R.; Chen, M.; Wang, Y. Chinese herbal medicine-derived compounds for cancer therapy: A focus on hepatocellular carcinoma. J. Ethnopharmacol. 2013, 149, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Wang, X.; Dang, Z.; Yu, L.; Wang, X.; Jiang, Y.; Yang, Z. Effects of adjuvant traditional Chinese medicine therapy on long-term survival in patients with hepatocellular carcinoma. Phytomed. Int. J. Phytother. Phytopharm. 2019, 62, 152930. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Dugoua, J.J.; Eyawo, O.; Mills, E.J. Traditional Chinese Medicines in the treatment of hepatocellular cancers: A systematic review and meta-analysis. J. Exp. Clin. Cancer Res. 2009, 28, 112. [Google Scholar] [CrossRef] [Green Version]
- Rajakrishnan, R.; Lekshmi, R.; Benil, P.B.; Thomas, J.; AlFarhan, A.H.; Rakesh, V.; Khalaf, S. Phytochemical evaluation of roots of Plumbago zeylanica L. and assessment of its potential as a nephroprotective agent. Saudi J. Biol. Sci. 2017, 24, 760–766. [Google Scholar] [CrossRef]
- Hassan, S.T.; Berchová-Bímová, K.; Petráš, J. Plumbagin, a Plant-Derived Compound, Exhibits Antifungal Combinatory Effect with Amphotericin B against Candida albicans Clinical Isolates and Anti-hepatitis C Virus Activity. Phytother. Res. PTR 2016, 30, 1487–1492. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Panda, M.; Biswal, B.K. Emerging role of plumbagin: Cytotoxic potential and pharmaceutical relevance towards cancer therapy. Food Chem. Toxicol. 2019, 125, 566–582. [Google Scholar] [CrossRef]
- Nair, S.V.; Baranwal, G.; Chatterjee, M.; Sachu, A.; Vasudevan, A.K.; Bose, C.; Banerji, A.; Biswas, R. Antimicrobial activity of plumbagin, a naturally occurring naphthoquinone from Plumbago rosea, against Staphylococcus aureus and Candida albicans. Int. J. Med. Microbiol. IJMM 2016, 306, 237–248. [Google Scholar] [CrossRef]
- Yu, T.; Xu, Y.Y.; Zhang, Y.Y.; Li, K.Y.; Shao, Y.; Liu, G. Plumbagin suppresses the human large cell lung cancer cell lines by inhibiting IL-6/STAT3 signaling in vitro. Int. Immunopharmacol. 2018, 55, 290–296. [Google Scholar] [CrossRef]
- Pradubyat, N.; Sakunrangsit, N.; Mutirangura, A.; Ketchart, W. NADPH: Quinone oxidoreductase 1 (NQO1) mediated anti-cancer effects of plumbagin in endocrine resistant MCF7 breast cancer cells. Phytomed. Int. J. Phytother. Phytopharm. 2020, 66, 153133. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Zhong, W.; Mustafa, A.; Fischer, J.W.; Witkowsky, O.; Verma, A.K. Plumbagin inhibits prostate cancer development in TRAMP mice via targeting PKCε, Stat3 and neuroendocrine markers. Carcinogenesis 2012, 33, 2586–2592. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cai, Y.; He, C.; Chen, M.; Li, H. Anticancer Properties and Pharmaceutical Applications of Plumbagin: A Review. Am. J. Chin. Med. 2017, 45, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, Y.; Wang, S.; Ma, J.; Peng, Y.; Yuan, X.; Lv, B.; Chen, W.; Wei, Y. Plumbagin induces autophagy and apoptosis of SMMC-7721 cells in vitro and in vivo. J. Cell Biochem. 2019, 120, 9820–9830. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lv, B.; Xie, J.; Zhang, Y.; Lin, Y.; Wang, S.; Zhong, J.; Chen, Y.; Peng, Y.; Ma, J. Plumbagin promotes human hepatoma SMMC-7721 cell apoptosis via caspase-3/vimentin signal-mediated EMT. Drug Des. Devel. Ther. 2019, 13, 2343–2355. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Lin, Y.; Chen, W.; Liu, S.; Jin, L.; Huang, D. Computational and In Vitro Analysis of Plumbagin’s Molecular Mechanism for the Treatment of Hepatocellular Carcinoma. Front. Pharmacol. 2021, 12, 594833. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis Int. J. Program. Cell Death 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Vaz Crippa, G.; Zanetti, T.A.; Biazi, B.I.; Baranoski, A.; Marques, L.A.; Coatti, G.C.; Lepri, S.R.; Mantovani, M.S. Up and down-regulation of mRNA in the cytotoxicity and genotoxicity of Plumbagin in HepG2/C3A. Environ. Toxicol. Pharmacol. 2020, 75, 103328. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.H.; Ryu, J.M.; Jeon, Y.J.; Choi, J.; Han, H.J.; Lee, Y.M.; Lee, S.; Bae, J.S.; Jung, J.W.; Chang, W.; et al. The role of thioredoxin reductase and glutathione reductase in plumbagin-induced, reactive oxygen species-mediated apoptosis in cancer cell lines. Eur. J. Pharmacol. 2015, 765, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Lv, M.; Chen, X.; Yu, Y.; Zang, G.; Tang, Z. Plumbagin inhibits proliferation and induces apoptosis of hepatocellular carcinoma by downregulating the expression of SIVA. Drug Des. Dev. Ther. 2019, 13, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Reviews. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.C.; Chiang, Y.M.; Sung, S.C.; Hsu, Y.L.; Chang, J.K.; Kuo, P.L. Plumbagin induces cell cycle arrest and apoptosis through reactive oxygen species/c-Jun N-terminal kinase pathways in human melanoma A375.S2 cells. Cancer Lett. 2008, 259, 82–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kan, H.; Liu, Y.; Ding, W. Plumbagin induces Ishikawa cell cycle arrest, autophagy, and apoptosis via the PI3K/Akt signaling pathway in endometrial cancer. Food Chem. Toxicol. 2021, 148, 111957. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Q.; Zhou, Z.W.; Yu, S.N.; Pan, S.T.; He, Z.X.; Zhang, X.; Wang, D.; Yang, Y.X.; Yang, T.; et al. Plumbagin induces cell cycle arrest and autophagy and suppresses epithelial to mesenchymal transition involving PI3K/Akt/mTOR-mediated pathway in human pancreatic cancer cells. Drug Des. Devel. Ther. 2015, 9, 537–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, A.; Sabarwal, A.; Narayan Mishra, J.P.; Singh, R.P. Plumbagin induces ROS-mediated apoptosis and cell cycle arrest and inhibits EMT in human cervical carcinoma cells. RSC Adv. 2018, 8, 32022–32037. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, D.; Yu, K.; Hu, Y.; Wu, Q.; Qian, F.; Wang, Z. CMPD1 inhibited human gastric cancer cell proliferation by inducing apoptosis and G2/M cell cycle arrest. Biol. Res. 2018, 51, 11. [Google Scholar] [CrossRef] [Green Version]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.K.; Rengasamy, K.R.R.; Biswal, B.K. Plumbagin engenders apoptosis in lung cancer cells via caspase-9 activation and targeting mitochondrial-mediated ROS induction. Arch. Pharmacal Res. 2020, 43, 242–256. [Google Scholar] [CrossRef]
- Sameni, S.; Hande, M.P. Plumbagin triggers DNA damage response, telomere dysfunction and genome instability of human breast cancer cells. Biomed. Pharmacother. 2016, 82, 256–268. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.L.; Lin, J.Y.; Shieh, J.C.; Yeh, H.F.; Hsieh, Y.H.; Cheng, Y.C.; Lee, H.J.; Shen, C.Y.; Cheng, C.W. Induction of G2/M Phase Arrest by Diosgenin via Activation of Chk1 Kinase and Cdc25C Regulatory Pathways to Promote Apoptosis in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 21, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Qu, J.; Gao, Z.; Qi, Q.; Yin, H.; Zhu, L.; Wu, Y.; Liu, W.; Yang, J.; Huang, X. Timosaponin AIII Induces G2/M Arrest and Apoptosis in Breast Cancer by Activating the ATM/Chk2 and p38 MAPK Signaling Pathways. Front. Pharmacol. 2020, 11, 601468. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.L.; Hsu, Y.L.; Cho, C.Y. Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol. Cancer Ther. 2006, 5, 3209–3221. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yeon, J.H.; Kim, H.; Roh, W.; Chae, J.; Park, H.O.; Kim, D.M. The natural anticancer agent plumbagin induces potent cytotoxicity in MCF-7 human breast cancer cells by inhibiting a PI-5 kinase for ROS generation. PLoS ONE 2012, 7, e45023. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Carlsen, L.; El-Deiry, W.S. Differential p53-Mediated Cellular Responses to DNA-Damaging Therapeutic Agents. Int. J. Mol. Sci. 2021, 22, 11828. [Google Scholar] [CrossRef] [PubMed]
- De, U.; Son, J.Y.; Jeon, Y.; Ha, S.Y.; Park, Y.J.; Yoon, S.; Ha, K.T.; Choi, W.S.; Lee, B.M.; Kim, I.S.; et al. Plumbagin from a tropical pitcher plant (Nepenthes alata Blanco) induces apoptotic cell death via a p53-dependent pathway in MCF-7 human breast cancer cells. Food Chem. Toxicol. 2019, 123, 492–500. [Google Scholar] [CrossRef]
- Tian, L.; Yin, D.; Ren, Y.; Gong, C.; Chen, A.; Guo, F.J. Plumbagin induces apoptosis via the p53 pathway and generation of reactive oxygen species in human osteosarcoma cells. Mol. Med. Rep. 2012, 5, 126–132. [Google Scholar] [CrossRef]
- Zhou, R.; Wu, K.; Su, M.; Li, R. Bioinformatic and experimental data decipher the pharmacological targets and mechanisms of plumbagin against hepatocellular carcinoma. Environ. Toxicol. Pharmacol. 2019, 70, 103200. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Herman-Antosiewicz, A.; Antosiewicz, J.; Xiao, H.; Brisson, M.; Lazo, J.S.; Singh, S.V. Diallyl trisulfide-induced G(2)-M phase cell cycle arrest in human prostate cancer cells is caused by reactive oxygen species-dependent destruction and hyperphosphorylation of Cdc 25 C. Oncogene 2005, 24, 6256–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gac, G.; Estève, P.O.; Ferec, C.; Pradhan, S. DNA damage-induced down-regulation of human Cdc25C and Cdc2 is mediated by cooperation between p53 and maintenance DNA (cytosine-5) methyltransferase 1. J. Biol. Chem. 2006, 281, 24161–24170. [Google Scholar] [CrossRef] [Green Version]
- St Clair, S.; Giono, L.; Varmeh-Ziaie, S.; Resnick-Silverman, L.; Liu, W.J.; Padi, A.; Dastidar, J.; DaCosta, A.; Mattia, M.; Manfredi, J.J. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: One involves direct binding to the cdc25C promoter. Mol. Cell 2004, 16, 725–736. [Google Scholar] [CrossRef]
- Bello, I.J.; Oyebode, O.T.; Olanlokun, J.O.; Omodara, T.O.; Olorunsogo, O.O. Plumbagin induces testicular damage via mitochondrial-dependent cell death. Chem. Biol. Interact. 2021, 347, 109582. [Google Scholar] [CrossRef]
- Gowda, R.; Kardos, G.; Sharma, A.; Singh, S.; Robertson, G.P. Nanoparticle-Based Celecoxib and Plumbagin for the Synergistic Treatment of Melanoma. Mol. Cancer Ther. 2017, 16, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Sakpakdeejaroen, I.; Somani, S.; Laskar, P.; Irving, C.; Mullin, M.; Dufès, C. Anti-Tumor Activity of Intravenously Administered Plumbagin Entrapped in Targeted Nanoparticles. J. Biomed. Nanotechnol. 2020, 16, 85–100. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Zhang, W.; Jin, L.; Liu, S.; Liang, L.; Wei, Y. Plumbagin Exhibits Genotoxicity and Induces G2/M Cell Cycle Arrest via ROS-Mediated Oxidative Stress and Activation of ATM-p53 Signaling Pathway in Hepatocellular Cells. Int. J. Mol. Sci. 2023, 24, 6279. https://doi.org/10.3390/ijms24076279
Liu H, Zhang W, Jin L, Liu S, Liang L, Wei Y. Plumbagin Exhibits Genotoxicity and Induces G2/M Cell Cycle Arrest via ROS-Mediated Oxidative Stress and Activation of ATM-p53 Signaling Pathway in Hepatocellular Cells. International Journal of Molecular Sciences. 2023; 24(7):6279. https://doi.org/10.3390/ijms24076279
Chicago/Turabian StyleLiu, Huan, Wenchao Zhang, Lijie Jin, Shasha Liu, Liying Liang, and Yanfei Wei. 2023. "Plumbagin Exhibits Genotoxicity and Induces G2/M Cell Cycle Arrest via ROS-Mediated Oxidative Stress and Activation of ATM-p53 Signaling Pathway in Hepatocellular Cells" International Journal of Molecular Sciences 24, no. 7: 6279. https://doi.org/10.3390/ijms24076279
APA StyleLiu, H., Zhang, W., Jin, L., Liu, S., Liang, L., & Wei, Y. (2023). Plumbagin Exhibits Genotoxicity and Induces G2/M Cell Cycle Arrest via ROS-Mediated Oxidative Stress and Activation of ATM-p53 Signaling Pathway in Hepatocellular Cells. International Journal of Molecular Sciences, 24(7), 6279. https://doi.org/10.3390/ijms24076279