
Smooth Muscle Cell Phenotypic Switch Induced by Traditional Cigarette Smoke Condensate: A Holistic Overview

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
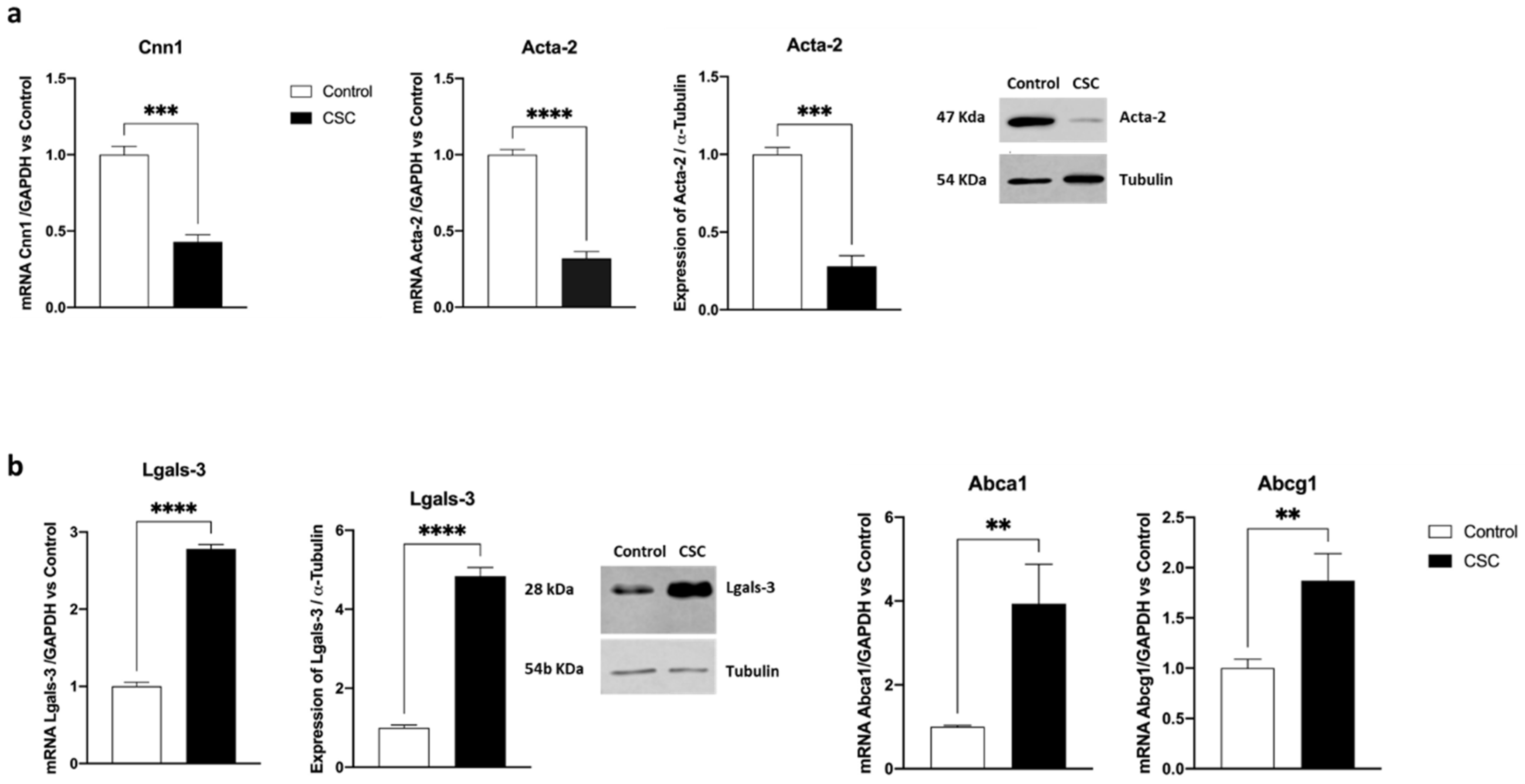
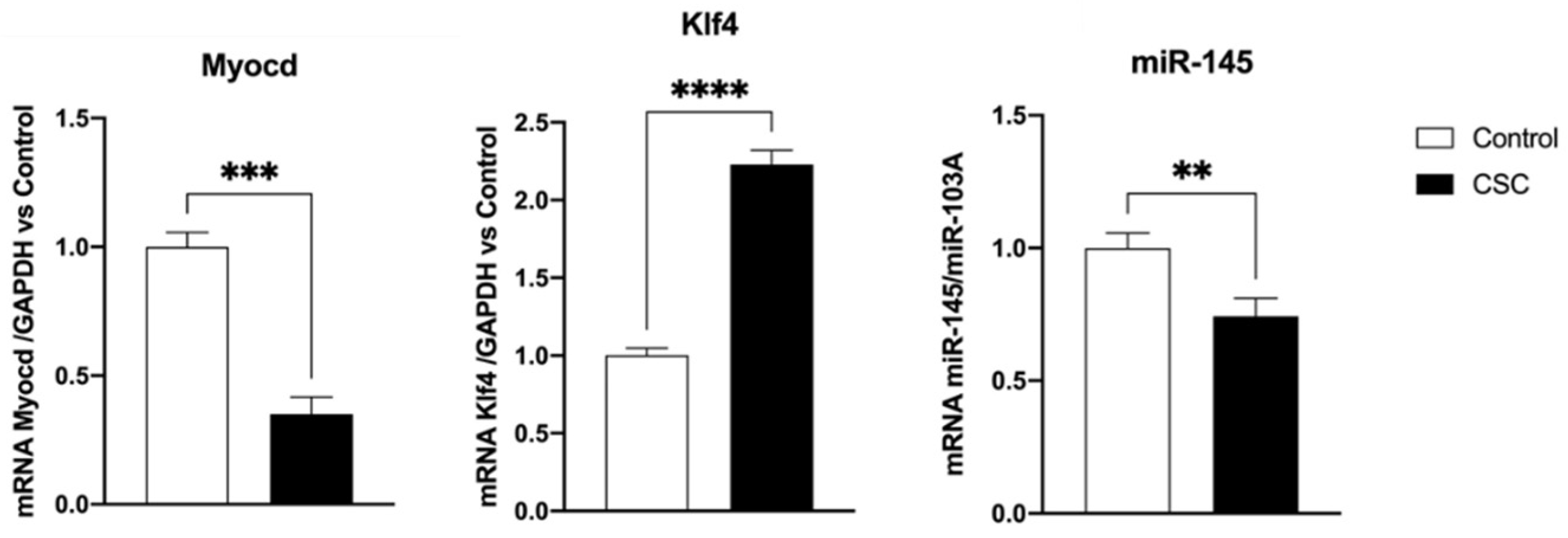
2.1. Effect of Cigarette Smoke Condensate on Murine SMC Phenotypic Switch
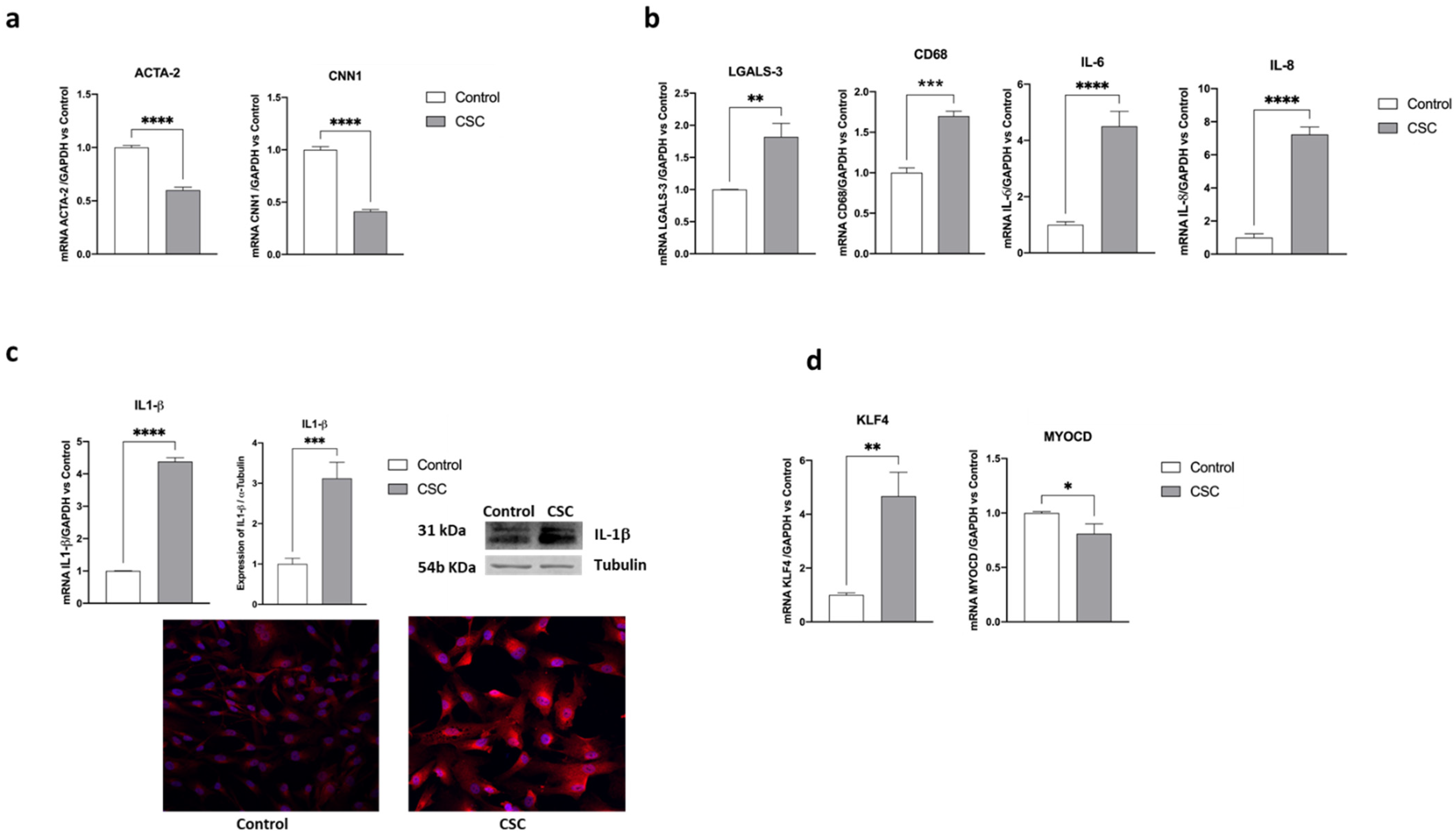
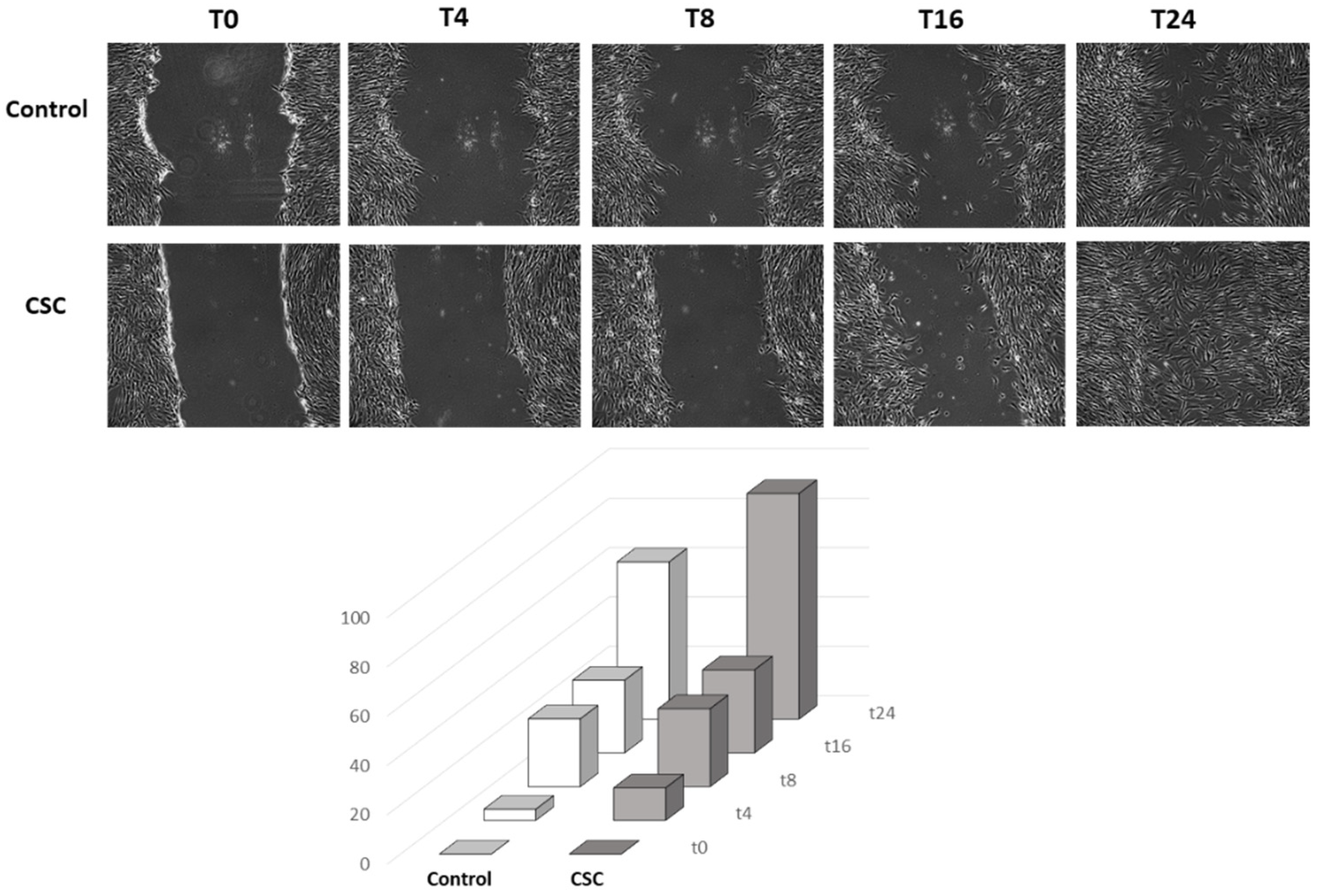
2.2. Effect of Cigarette Smoke Condensate on Human SMC Phenotypic Switch
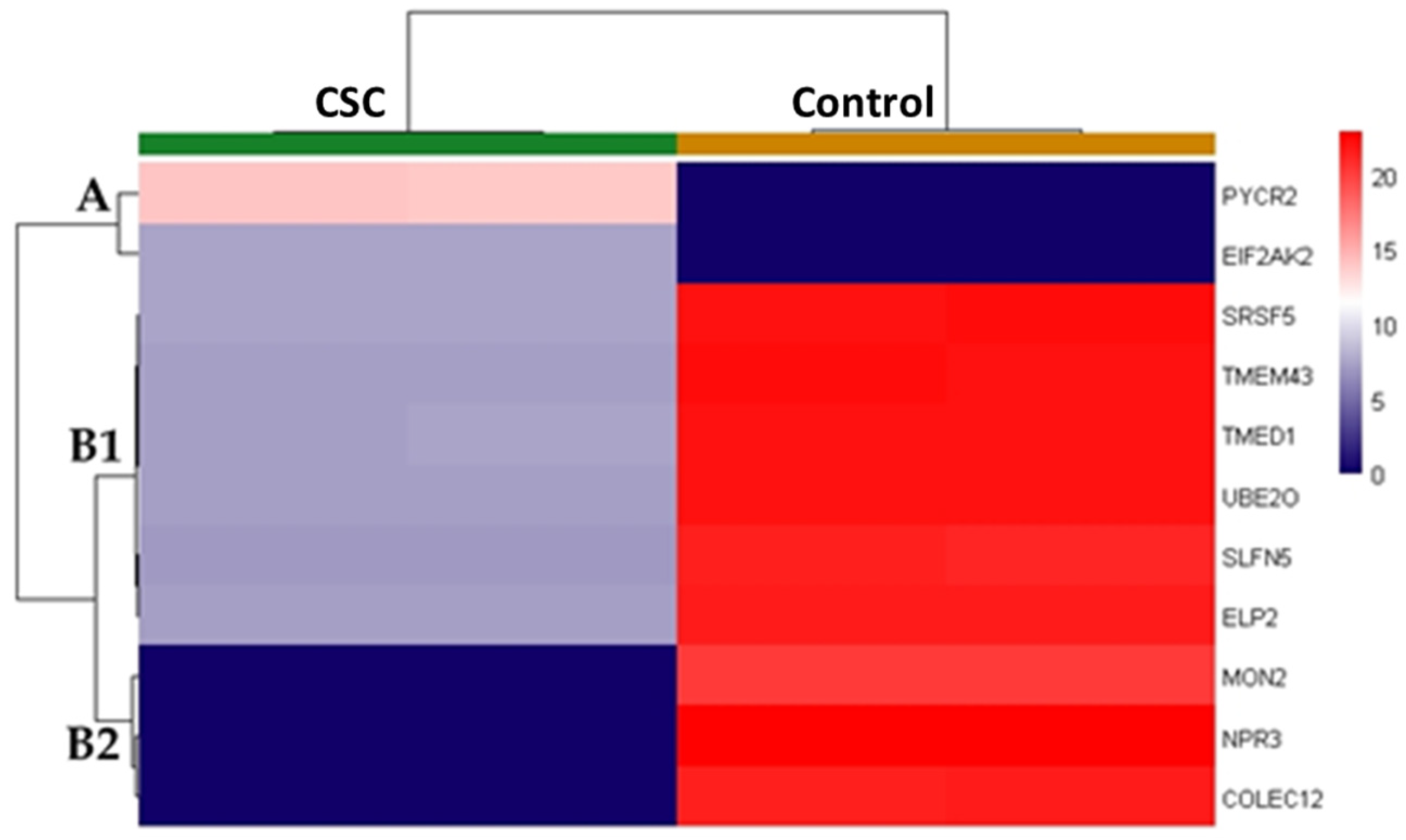
2.3. CSC Effects on VSMC Proteomic Profile and Combined Functional Analyses of Proteins and Genes Significantly Deregulated by the CSC Treatment
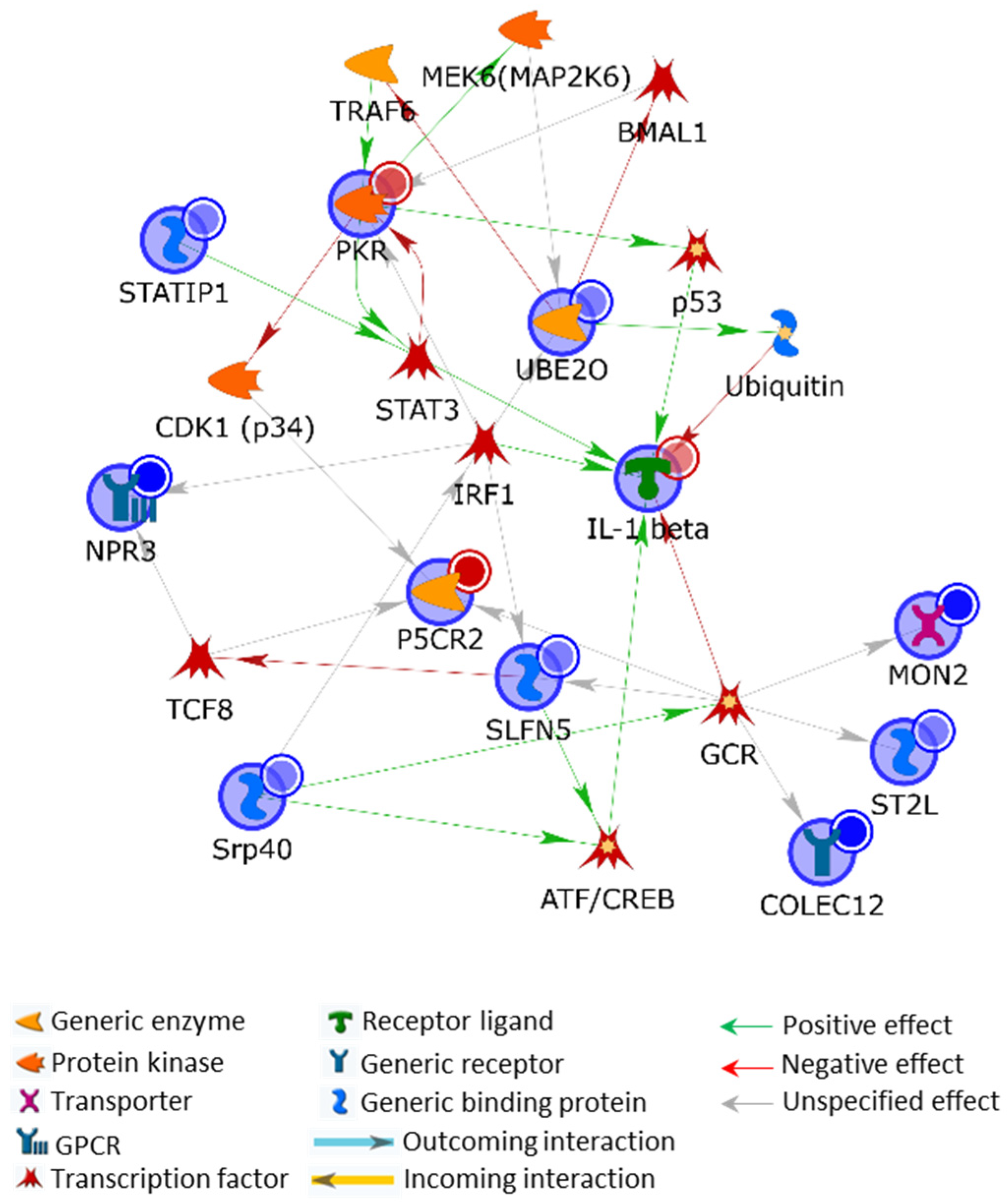
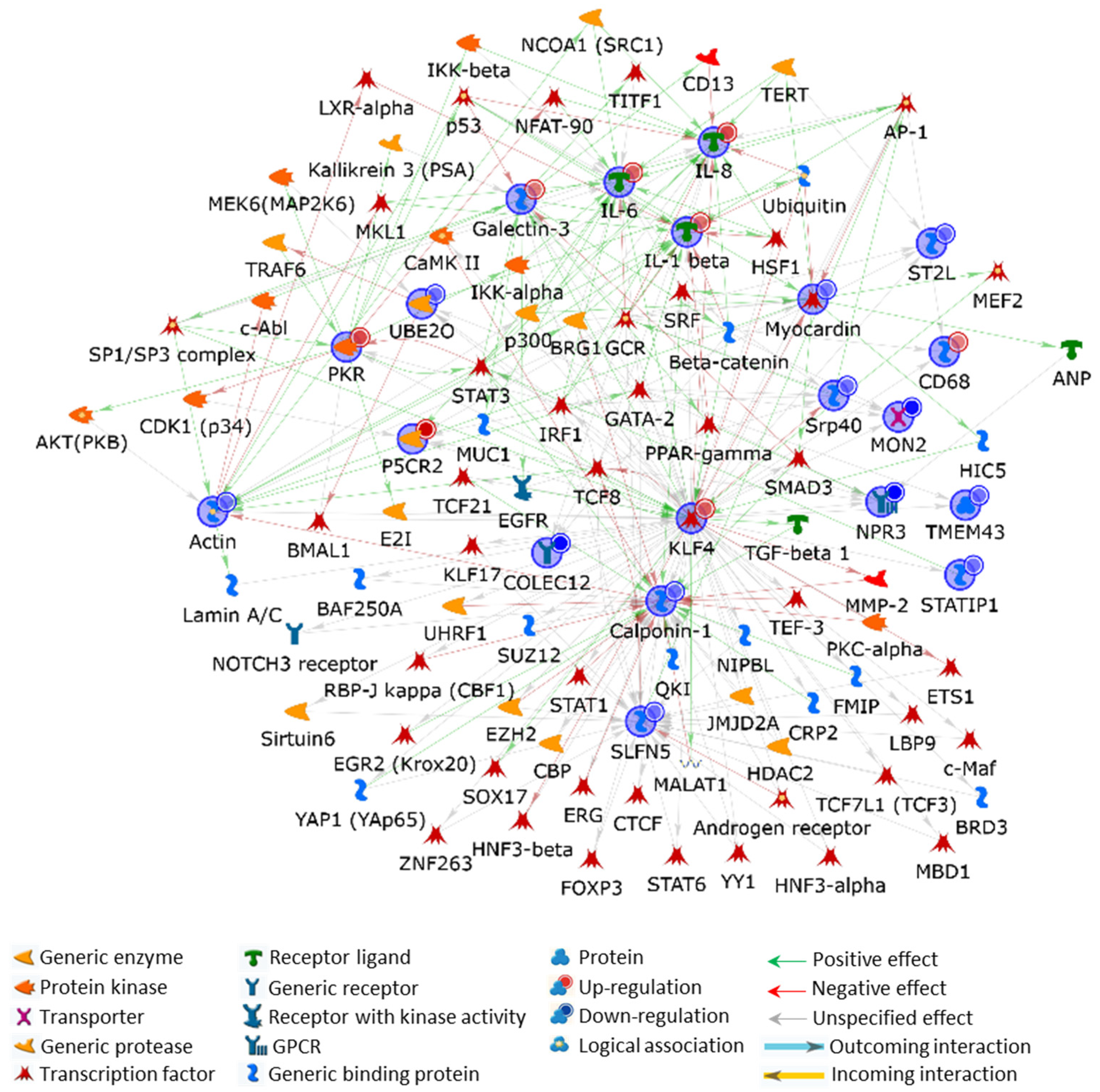
2.3.1. Protein Hybrid Network
2.3.2. Protein/Gene Hybrid Network
3. Materials and Methods
3.1. Cell Culture
3.2. Cigarette Smoke Condensate
3.3. Confocal Microscopy
3.4. RNA Isolation and Reverse Transcription
3.5. Quantitative RT-PCR
3.6. PCR Arrays
3.7. miRNA Expression
3.8. Protein Isolation, Quantification, SDS-PAGE and Western Blot
3.9. Cell Proliferation
3.10. In Vitro Directional Migration (Wound Healing Assay)
3.11. Comparative Mass Spectrometry Proteomics
3.12. Functional Analysis of Detected Differences in the CSC vs. Control Comparison
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Klein, L.W. Pathophysiologic Mechanisms of Tobacco Smoke Producing Atherosclerosis. Curr. Cardiol. Rev. 2022, 18. [Google Scholar] [CrossRef] [PubMed]
- Golbidi, S.; Edvinsson, L.; Laher, I. Smoking and Endothelial Dysfunction. Curr. Vasc. Pharmacol. 2019, 18, 1–11. [Google Scholar] [CrossRef]
- Barbieri, S.S.; Weksler, B.B. Tobacco Smoke Cooperates with Interleukin-1beta to Alter Beta-Catenin Trafficking in Vascular Endothelium Resulting in Increased Permeability and Induction of Cyclooxygenase-2 Expression in Vitro and in Vivo. FASEB J. 2007, 21, 1831–1843. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.; Mieremet, A.; de Vries, C.J.M.; Micha, D.; de Waard, V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2693–2707. [Google Scholar] [CrossRef]
- Gomez, D.; Owens, G.K. Smooth Muscle Cell Phenotypic Switching in Atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol Loading Reprograms the MicroRNA-143/145-Myocardin Axis to Convert Aortic Smooth Muscle Cells to a Dysfunctional Macrophage-Like Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of Mouse Aortic Smooth Muscle Cells to a Macrophage-like State after Cholesterol Loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, S.; Monti, M.; Arnaboldi, L.; Canavesi, M.; Ainis Buscherini, G.; Calabresi, L.; Corsini, A.; Bellosta, S. ABCA1 and HDL 3 Are Required to Modulate Smooth Muscle Cells Phenotypic Switch after Cholesterol Loading. Atherosclerosis 2017, 266, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.M.; Ali, M.S.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, F.; Hasan, D.M.; Rosenwasser, R.H.; Owens, G.K.; Koch, W.J.; Dumont, A.S. Cigarette Smoke Modulates Vascular Smooth Muscle Phenotype: Implications for Carotid and Cerebrovascular Disease. PLoS ONE 2013, 8, e71954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth Muscle Cell Fate and Plasticity in Atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Xiang, Y.; Fan, L.J.; Zhang, X.Y.; Li, J.P.; Yu, C.X.; Bao, L.Y.; Cao, D.S.; Xing, W.B.; Liao, X.H.; et al. Myocardin Inhibited the Gap Protein Connexin 43 via Promoted MiR-206 to Regulate Vascular Smooth Muscle Cell Phenotypic Switch. Gene 2017, 616, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Ackers-Johnson, M.; Talasila, A.; Sage, A.P.; Long, X.; Bot, I.; Morrell, N.W.; Bennett, M.R.; Miano, J.M.; Sinha, S. Myocardin Regulates Vascular Smooth Muscle Cell Inflammatory Activation and Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 817–828. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Gomez, D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1715–1723. [Google Scholar] [CrossRef]
- Deaton, R.A.; Gan, Q.; Owens, G.K. Sp1-Dependent Activation of KLF4 Is Required for PDGF-BB-Induced Phenotypic Modulation of Smooth Muscle. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1027–H1037. [Google Scholar] [CrossRef] [Green Version]
- Bulut, G.B.; Alencar, G.F.; Owsiany, K.M.; Nguyen, A.T.; Karnewar, S.; Haskins, R.M.; Waller, L.K.; Cherepanova, O.A.; Deaton, R.A.; Shankman, L.S.; et al. KLF4 (Kruppel-Like Factor 4)-Dependent Perivascular Plasticity Contributes to Adipose Tissue Inflammation. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 284–301. [Google Scholar] [CrossRef]
- Boettger, T.; Beetz, N.; Kostin, S.; Schneider, J.; Krüger, M.; Hein, L.; Braun, T. Acquisition of the Contractile Phenotype by Murine Arterial Smooth Muscle Cells Depends on the Mir143/145 Gene Cluster. J. Clin. Investig. 2009, 119, 2634–2647. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Li, B.; Xu, Y.; Yang, P.; Chen, R.; Wang, Z.; Shao, C.; Song, J.; Yan, J. Hypermethylation of the Micro-RNA 145 Promoter Is the Key Regulator for NLRP3 Inflammasome-Induced Activation and Plaque Formation. JACC Basic Transl. Sci. 2018, 3, 604–624. [Google Scholar] [CrossRef]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.-H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. MiR-145 and MiR-143 Regulate Smooth Muscle Cell Fate and Plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chen, L.; Shang, C.; Jin, Z.; Yao, F.; Bai, L.; Wang, R.; Zhao, S.; Liu, E. MiR-145 Inhibits the Proliferation and Migration of Vascular Smooth Muscle Cells by Regulating Autophagy. J. Cell Mol. Med. 2020, 24, 6658–6669. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; Massy, Z.A.; Meuth, V.M.-L.; Metzinger, L. MiR-143 and MiR-145 Molecular Keys to Switch the Phenotype of Vascular Smooth Muscle Cells. Circ. Cardiovasc. Genet. 2011, 4, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, S.; Monti, M.; Buscherini, G.A.; Arnaboldi, L.; Canavesi, M.; Corsini, A.; Bellosta, S. The Dataset Describes: Phenotypic Changes Induced by Cholesterol Loading in Smooth Muscle Cells Isolated from the Aortae of C57BL/6 Mice. Data Brief 2018, 16, 334–340. [Google Scholar] [CrossRef]
- Blalock, W.L.; Piazzi, M.; Bavelloni, A.; Raffini, M.; Faenza, I.; D’Angelo, A.; Cocco, L. Identification of the PKR Nuclear Interactome Reveals Roles in Ribosome Biogenesis, MRNA Processing and Cell Division. J. Cell Physiol. 2014, 229, 1047–1060. [Google Scholar] [CrossRef]
- Mao, Z.; Liu, C.; Lin, X.; Sun, B.; Su, C. PPP2R5A: A Multirole Protein Phosphatase Subunit in Regulating Cancer Development. Cancer Lett. 2018, 414, 222–229. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, Y.; Wu, Y.; Kumar, V.; Mahato, R.I.; Su, Q. Activation of DsRNA-Dependent Protein Kinase R by MiR-378 Sustains Metabolic Inflammation in Hepatic Insulin Resistance. Diabetes 2021, 70, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jee, H.-Y.; Lee, Y.-G.; Shin, J.-I.; Jeon, Y.-J.; Kim, J.-B.; Seo, H.; Lee, J.-Y.; Lee, K. PKR-Mediated Phosphorylation of EIF2a and CHK1 Is Associated with Doxorubicin-Mediated Apoptosis in HCC1143 Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 15872. [Google Scholar] [CrossRef] [PubMed]
- Chukwurah, E.; Farabaugh, K.T.; Guan, B.; Ramakrishnan, P.; Hatzoglou, M. A Tale of Two Proteins: PACT and PKR and Their Roles in Inflammation. FEBS J. 2021, 288, 6365–6391. [Google Scholar] [CrossRef] [PubMed]
- Kalra, J.; Mangali, S.; Bhat, A.; Jadhav, K.; Dhar, A. Selective Inhibition of PKR Improves Vascular Inflammation and Remodelling in High Fructose Treated Primary Vascular Smooth Muscle Cells. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165606. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ribeiro, D.; Godinas, L.; Pilette, C.; Perros, F. The Integrated Stress Response System in Cardiovascular Disease. Drug Discov. Today 2018, 23, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, L.; Tu, M.; Huang, M.; Chen, Y.; Pan, D.; Peng, J.; Shen, X. The Ameliorative Effect of Terpinen-4-Ol on ER Stress-Induced Vascular Calcification Depends on SIRT1-Mediated Regulation of PERK Acetylation. Pharmacol. Res. 2021, 170, 105629. [Google Scholar] [CrossRef]
- Das, S.; Ward, S.V.; Tacke, R.S.; Suske, G.; Samuel, C.E. Activation of the RNA-Dependent Protein Kinase PKR Promoter in the Absence of Interferon Is Dependent Upon Sp Proteins. J. Biol. Chem. 2006, 281, 3244–3253. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.; Ande, A.; Sinha, N.; Kumar, A.; Kumar, S. Effects of Cigarette Smoke Condensate on Oxidative Stress, Apoptotic Cell Death, and HIV Replication in Human Monocytic Cells. PLoS ONE 2016, 11, e0155791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellamri, M.; Walmsley, S.J.; Brown, C.; Brandt, K.; Konorev, D.; Day, A.; Wu, C.-F.; Wu, M.T.; Turesky, R.J. DNA Damage and Oxidative Stress of Tobacco Smoke Condensate in Human Bladder Epithelial Cells. Chem. Res. Toxicol. 2022, 35, 1863–1880. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, L.; Chen, X.; Liang, M.L.; Wei, D.H.; Cao, J.; Zhang, J. Cigarette Smoke Extract Stimulates Human Pulmonary Artery Smooth Muscle Cell Proliferation: Role of Inflammation and Oxidative Stress. Iran J. Basic Med. Sci. 2022, 25, 755–761. [Google Scholar] [CrossRef]
- Starke, R.M.; Thompson, J.W.; Ali, M.S.; Pascale, C.L.; Lege, A.M.; Ding, D.; Chalouhi, N.; Hasan, D.M.; Jabbour, P.; Owens, G.K.; et al. Cigarette Smoke Initiates Oxidative Stress-Induced Cellular Phenotypic Modulation Leading to Cerebral Aneurysm Pathogenesis. Arterioscler. Thromb. Vasc. Biol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Somborac-Bačura, A.; van der Toorn, M.; Franciosi, L.; Slebos, D.-J.; Žanić-Grubišić, T.; Bischoff, R.; van Oosterhout, A.J.M. Cigarette Smoke Induces Endoplasmic Reticulum Stress Response and Proteasomal Dysfunction in Human Alveolar Epithelial Cells. Exp. Physiol. 2013, 98, 316–325. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel Role of PKR in Inflammasome Activation and HMGB1 Release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [Green Version]
- Carvalho-Filho, M.A.; Carvalho, B.M.; Oliveira, A.G.; Guadagnini, D.; Ueno, M.; Dias, M.M.; Tsukumo, D.M.; Hirabara, S.M.; Reis, L.F.; Curi, R.; et al. Double-Stranded RNA-Activated Protein Kinase Is a Key Modulator of Insulin Sensitivity in Physiological Conditions and in Obesity in Mice. Endocrinology 2012, 153, 5261–5274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sud, N.; Rutledge, A.; Pan, K.; Su, Q. Activation of the DsRNA-Activated Protein Kinase PKR in Mitochondrial Dysfunction and Inflammatory Stress in Metabolic Syndrome. Curr. Pharm. Des. 2016, 22, 2697–2703. [Google Scholar] [CrossRef]
- Gilbert, S.J.; Duance, V.C.; Mason, D.J. Does Protein Kinase R Mediate TNF-Alpha- and Ceramide-Induced Increases in Expression and Activation of Matrix Metalloproteinases in Articular Cartilage by a Novel Mechanism? Arthritis Res. Ther. 2004, 6, R46–R55. [Google Scholar] [CrossRef] [Green Version]
- Deb, A. Protein Kinase PKR Is Required for Platelet-Derived Growth Factor Signaling of c-Fos Gene Expression via Erks and Stat3. EMBO J. 2001, 20, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.-H.; Wang, N.; Zhao, D.-W.; Zheng, D.-L.; Zheng, L.; Xing, W.-J.; Ma, W.-J.; Bao, L.-Y.; Dong, J.; Zhang, T.-C. STAT3 Protein Regulates Vascular Smooth Muscle Cell Phenotypic Switch by Interaction with Myocardin. J. Biol. Chem. 2015, 290, 19641–19652. [Google Scholar] [CrossRef] [Green Version]
- Collum, R.G.; Brutsaert, S.; Lee, G.; Schindler, C. A Stat3-Interacting Protein (StIP1) Regulates Cytokine Signal Transduction. Proc. Natl. Acad. Sci. USA 2000, 97, 10120–10125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.; Hall, S.; Lanford, H.; Ward, N.; Grespin, R.T.; Figueroa, M.; Mattia, V.; Xiong, Y.; Mukherjee, R.; Jones, J.; et al. Signaling through the IL-6-STAT3 Pathway Promotes Proteolytically-Active Macrophage Accumulation Necessary for Development of Small AAA. Vasc. Endovasc. Surg. 2023, 153857442311529. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Malik, R.; Richardson, T.G.; Howson, J.M.M.; Anderson, C.D.; Burgess, S.; Hovingh, G.K.; Dichgans, M.; Gill, D. Associations of Genetically Predicted IL-6 Signaling with Cardiovascular Disease Risk across Population Subgroups. BMC Med. 2022, 20. [Google Scholar] [CrossRef]
- Xu, C.-P.; Li, X.; Hu, Y.-J.; Cui, Z.; Wang, L.; Liang, L.; Zhou, Y.-L.; Yang, Y.-J.; Yu, B. Quantitative Proteomics Reveals ELP2 as a Regulator to the Inhibitory Effect of TNF-α on Osteoblast Differentiation. J. Proteomics. 2015, 114, 234–246. [Google Scholar] [CrossRef]
- Baek, M.; Yoo, E.; Choi, H.I.; An, G.Y.; Chai, J.C.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. The BET Inhibitor Attenuates the Inflammatory Response and Cell Migration in Human Microglial HMC3 Cell Line. Sci. Rep. 2021, 11, 8828. [Google Scholar] [CrossRef]
- Buccione, C.; Fragale, A.; Polverino, F.; Ziccheddu, G.; Aricò, E.; Belardelli, F.; Proietti, E.; Battistini, A.; Moschella, F. Role of interferon regulatory factor 1 in governing Treg depletion, Th1 polarization, inflammasome activation and antitumor efficacy of cyclophosphamide. Int. J. Cancer 2018, 142, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruckmeier, M.; Kuehnl, A.; Culmes, M.; Pelisek, J.; Eckstein, H.-H. Impact of OxLDL and LPS on C-Type Natriuretic Peptide System Is Different between THP-1 Cells and Human Peripheral Blood Monocytic Cells. Cell. Physiol. Biochem. 2012, 30, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egom, E.E.-A. Pulmonary Arterial Hypertension Due to NPR-C Mutation: A Novel Paradigm for Normal and Pathologic Remodeling? Int. J. Mol. Sci. 2019, 20, 3063. [Google Scholar] [CrossRef] [Green Version]
- Moyes, A.J.; Khambata, R.S.; Villar, I.; Bubb, K.J.; Baliga, R.S.; Lumsden, N.G.; Xiao, F.; Gane, P.J.; Rebstock, A.-S.; Worthington, R.J.; et al. Endothelial C-Type Natriuretic Peptide Maintains Vascular Homeostasis. J. Clin. Investig. 2014, 124, 4039–4051. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shao, B.; Liu, Z.; Dang, Q.; Guo, Y.; Chen, C.; Guo, Y.; Chen, Z.; Liu, J.; Hu, S.; et al. LINC01296/MiR-141-3p/ZEB1-ZEB2 Axis Promotes Tumor Metastasis via Enhancing Epithelial-Mesenchymal Transition Process. J. Cancer 2021, 12, 2723–2734. [Google Scholar] [CrossRef]
- Sassano, A.; Mavrommatis, E.; Arslan, A.D.; Kroczynska, B.; Beauchamp, E.M.; Khuon, S.; Chew, T.-L.; Green, K.J.; Munshi, H.G.; Verma, A.K.; et al. Human Schlafen 5 (SLFN5) Is a Regulator of Motility and Invasiveness of Renal Cell Carcinoma Cells. Mol. Cell Biol. 2015, 35, 2684–2698. [Google Scholar] [CrossRef] [Green Version]
- Wan, G.; Liu, Y.; Zhu, J.; Guo, L.; Li, C.; Yang, Y.; Gu, X.; Deng, L.-L.; Lu, C. SLFN5 Suppresses Cancer Cell Migration and Invasion by Inhibiting MT1-MMP Expression via AKT/GSK-3β/β-Catenin Pathway. Cell Signal. 2019, 59, 1–12. [Google Scholar] [CrossRef]
- Takeyama, Y.; Sato, M.; Horio, M.; Hase, T.; Yoshida, K.; Yokoyama, T.; Nakashima, H.; Hashimoto, N.; Sekido, Y.; Gazdar, A.F.; et al. Knockdown of ZEB1, a Master Epithelial-to-Mesenchymal Transition (EMT) Gene, Suppresses Anchorage-Independent Cell Growth of Lung Cancer Cells. Cancer Lett. 2010, 296, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.; Liang, S.; Wang, Y.; Jiao, B. Knocking down TCF8 Inhibits High Glucose- and Angiotensin II-Induced Epithelial to Mesenchymal Transition in Podocytes. Biosci. Trends 2017, 11, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Wan, G.; Yang, Y.; Liu, Y.; Yang, X.; Zheng, Y.; Jiang, L.; Zhang, P.; Liu, D.; Zhao, W.; et al. SLFN5 Influences Proliferation and Apoptosis by Upregulating PTEN Transcription via ZEB1 and Inhibits the Purine Metabolic Pathway in Breast Cancer. Am. J. Cancer Res. 2020, 10, 2832–2850. [Google Scholar]
- Zhang, M.; Yang, C.; Ruan, X.; Liu, X.; Wang, D.; Liu, L.; Shao, L.; Wang, P.; Dong, W.; Xue, Y. CPEB2 M6A Methylation Regulates Blood–Tumor Barrier Permeability by Regulating Splicing Factor SRSF5 Stability. Commun. Biol. 2022, 5, 908. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Wordinger, R.J.; Yorio, T.; Clark, A.F. Spliceosome Protein (SRp) Regulation of Glucocorticoid Receptor Isoforms and Glucocorticoid Response in Human Trabecular Meshwork Cells. Investig. Opthalmology Vis. Sci. 2012, 53, 857. [Google Scholar] [CrossRef]
- MacLeod, C.; Hadoke, P.W.F.; Nixon, M. Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers? Int. J. Mol. Sci. 2021, 22, 7622. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Artan, M.; Kim, N.; Yeom, J.; Hwang, A.B.; Jeong, D.-E.; Altintas, Ö.; Seo, K.; Seo, M.; Lee, D.; et al. MON-2, a Golgi Protein, Mediates Autophagy-Dependent Longevity in Caenorhabditis elegans. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- McGough, I.J.; de Groot, R.E.A.; Jellett, A.P.; Betist, M.C.; Varandas, K.C.; Danson, C.M.; Heesom, K.J.; Korswagen, H.C.; Cullen, P.J. SNX3-Retromer Requires an Evolutionary Conserved MON2:DOPEY2:ATP9A Complex to Mediate Wntless Sorting and Wnt Secretion. Nat. Commun. 2018, 9, 3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; de Meyer, G.R.Y. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Molière, A.; Beer, K.B.; Wehman, A.M. Dopey Proteins Are Essential but Overlooked Regulators of Membrane Trafficking. J. Cell Sci. 2022, 135. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, Y.; Mou, Y.; Zhao, Y.; Blankesteijn, W.M.; Hall, J.L. A Role for the β-Catenin/T-Cell Factor Signaling Cascade in Vascular Remodeling. Circ. Res. 2002, 90, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Albanese, I.; Khan, K.; Barratt, B.; Al-Kindi, H.; Schwertani, A. Atherosclerotic Calcification: Wnt Is the Hint. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-Y.; Ma, T.-L.; Chen, K.-N.; Pang, Z.-Y.; Wang, H.; Huang, J.-M.; Qi, G.-B.; Wang, C.-Z.; Jiang, Z.-X.; Gong, L.-J.; et al. Accumulation of LDL/Ox-LDL in the Necrotic Region Participates in Osteonecrosis of the Femoral Head: A Pathological and in Vitro Study. Lipids Health Dis. 2021, 20, 167. [Google Scholar] [CrossRef]
- Selman, L.; Skjodt, K.; Nielsen, O.; Floridon, C.; Holmskov, U.; Hansen, S. Expression and Tissue Localization of Collectin Placenta 1 (CL-P1, SRCL) in Human Tissues. Mol. Immunol. 2008, 45, 3278–3288. [Google Scholar] [CrossRef]
- Ashraf, M.Z.; Sahu, A. Scavenger Receptors: A Key Player in Cardiovascular Diseases. Biomol. Concepts 2012, 3, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Cuthbert, G.A.; Shaik, F.; Harrison, M.A.; Ponnambalam, S.; Homer-Vanniasinkam, S. Scavenger Receptors as Biomarkers and Therapeutic Targets in Cardiovascular Disease. Cells 2020, 9, 2453. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Suzuki, Y.; Eda, S.; Kawai, T.; Kase, T.; Keshi, H.; Sakai, Y.; Fukuoh, A.; Sakamoto, T.; Itabe, H.; et al. The Membrane-Type Collectin CL-P1 Is a Scavenger Receptor on Vascular Endothelial Cells. J. Biol. Chem. 2001, 276, 44222–44228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, J.L.; Ozment, T.R.; Li, C.; Schweitzer, J.B.; Williams, D.L. Scavenger Receptor-A (CD204): A Two-Edged Sword in Health and Disease. Crit. Rev. Immunol. 2014, 34, 241–261. [Google Scholar] [CrossRef] [Green Version]
- Ullah, K.; Zubia, E.; Narayan, M.; Yang, J.; Xu, G. Diverse roles of the E2/E3 hybrid enzyme UBE2O in the regulation of protein ubiquitination, cellular functions, and disease onset. FEBS J. 2019, 286, 2018–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagitani, K.; Juszkiewicz, S.; Hegde, R.S. UBE2O Is a Quality Control Factor for Orphans of Multiprotein Complexes. Science 2017, 357, 472–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, J.; Zhang, L.; van Dam, H.; ten Dijke, P. UBE2O Negatively Regulates TRAF6-Mediated NF-ΚB Activation by Inhibiting TRAF6 Polyubiquitination. Cell Res. 2013, 23, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Lou, C.; Chen, S.; Zhang, Z.; Xu, Q. XIAP and PHB1 Regulate Anoikis through Competitive Binding to TRAF6. Mol. Cancer Res. 2023, 21, 127–139. [Google Scholar] [CrossRef]
- Brasier, A.R. The Nuclear Factor- B-Interleukin-6 Signalling Pathway Mediating Vascular Inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Wan, G.; Zhu, J.; Gu, X.; Yang, Y.; Liu, Y.; Wang, Z.; Zhao, Y.; Wu, H.; Huang, G.; Lu, C. Human Schlafen 5 Regulates Reversible Epithelial and Mesenchymal Transitions in Breast Cancer by Suppression of ZEB1 Transcription. Br. J. Cancer 2020, 123, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like Factor 4 (KLF4): What We Currently Know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How Vascular Smooth Muscle Cell Phenotype Switching Contributes to Vascular Disease. Cell Commun. Signal. 2022, 20. [Google Scholar] [CrossRef] [PubMed]
- Harman, J.L.; Jørgensen, H.F. The Role of Smooth Muscle Cells in Plaque Stability: Therapeutic Targeting Potential. Br. J. Pharmacol. 2019, 176, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Bandyopadhyaya, G.; Polumuri, S.K.; Chumsri, S.; Gade, P.; Kalvakolanu, D.v.; Ahmed, H. Nicotine Promotes Apoptosis Resistance of Breast Cancer Cells and Enrichment of Side Population Cells with Cancer Stem Cell-like Properties via a Signaling Cascade Involving Galectin-3, A9 Nicotinic Acetylcholine Receptor and STAT3. Breast Cancer Res. Treat. 2014, 145, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, M.C.; Weil, R.; Dam, E.; Hovanessian, A.G.; Meurs, E.F. PKR Stimulates NF-ΚB Irrespective of Its Kinase Function by Interacting with the IκB Kinase Complex. Mol. Cell Biol. 2000, 20, 4532–4542. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Liu, S.; Shi, Y.; Liu, N.; Chen, L.; Wang, X.; Xiao, D.; Liu, X.; Mao, C.; Jiang, Y.; et al. Activation of AhR with Nuclear IKKα Regulates Cancer Stem-like Properties in the Occurrence of Radioresistance. Cell Death Dis. 2018, 9, 490. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Meng, Q.; Zhang, Z.; Yi, T.; He, Y.; Zheng, J.; Lei, W. Aryl Hydrocarbon Receptor Pathway: Role, Regulation and Intervention in Atherosclerosis Therapy (Review). Mol. Med. Rep. 2019, 20, 4763–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyrieux, A.F.; Wilson, V.G. Sumoylation in Development and Differentiation. Adv. Exp. Med. Biol. 2017, 963, 197–214. [Google Scholar]
- Shen, Y.; Xu, L.; Yan, D.; Zhou, M.; Han, T.; Lu, C.; Tang, X.; Lin, C.; Qian, R.; Guo, D. BMAL1 Modulates Smooth Muscle Cells Phenotypic Switch towards Fibroblast-like Cells and Stabilizes Atherosclerotic Plaques by Upregulating YAP1. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166450. [Google Scholar] [CrossRef]
- Dyar, K.A.; Hubert, M.J.; Mir, A.A.; Ciciliot, S.; Lutter, D.; Greulich, F.; Quagliarini, F.; Kleinert, M.; Fischer, K.; Eichmann, T.O.; et al. Transcriptional Programming of Lipid and Amino Acid Metabolism by the Skeletal Muscle Circadian Clock. PLoS Biol. 2018, 16, e2005886. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.-F.; Zhou, Z.-Y.; Liu, Y.-Z.; Wu, L.; Nie, B.-B.; Huang, L.; Zhang, C. Role of Sp1 in Atherosclerosis. Mol. Biol. Rep. 2022, 49, 9893–9902. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.v.; Handy, I.; Goldsmith, T.; Patel, R.C. PACT, a Stress-Modulated Cellular Activator of Interferon-Induced Double-Stranded RNA-Activated Protein Kinase, PKR. J. Biol. Chem. 2000, 275, 37993–37998. [Google Scholar] [CrossRef] [Green Version]
- Hay, C.; Micko, C.; Prescott, M.F.; Liau, G.; Robinson, K.; de Leon, H. Differential Cell Cycle Progression Patterns of Infiltrating Leukocytes and Resident Cells After Balloon Injury of the Rat Carotid Artery. Arter. Thromb. Vasc. Biol. 2001, 21, 1948–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.W.; Hastings, N.E.; Blackman, B.R.; Wamhoff, B.R. Complex Regulation and Function of the Inflammatory Smooth Muscle Cell Phenotype in Atherosclerosis. J. Vasc. Res. 2010, 47, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, Z.; Hu, X.; Zhang, Y.; Zhang, S. Synthetic E-Selectin Prevents Postoperative Vascular Restenosis by Inhibiting Nuclear Factor ΚB in Rats. Mol. Med. Rep. 2018, 17, 5065–5073. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, M. Krüppel-like Factor 4-Mediated Smooth Muscle Cell Phenotype Switching to a Galectin-3 Positive Subclass Is a Detrimental Event in the Pathogenesis of Atherosclerotic Cardiovascular Disease. Noncoding RNA Investig. 2020, 4, 8. [Google Scholar] [CrossRef]
- Ma, J.; Yao, Y.; Wang, P.; Liu, Y.; Zhao, L.; Li, Z.; Li, Z.; Xue, Y. MiR-152 Functions as a Tumor Suppressor in Glioblastoma Stem Cells by Targeting Krüppel-like Factor 4. Cancer Lett. 2014, 355, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Li, M.; Li, W. Galectin-3 Inhibition Ameliorates Hypoxia-Induced Pulmonary Artery Hypertension. Mol. Med. Rep. 2017, 15, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a Novel Biomarker for Disease Diagnosis and a Target for Therapy (Review). Int. J. Mol. Med. 2017, 41, 599–614. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Huang, C.-K.; Ding, F.; Zhang, R. Galectin-3 Mediates Thrombin-Induced Vascular Smooth Muscle Cell Migration. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Tian, L.; Chen, K.; Cao, J.; Han, Z.; Wang, Y.; Gao, L.; Fan, Y.; Wang, C. Galectin-3 Induces the Phenotype Transformation of Human Vascular Smooth Muscle Cells via the Canonical Wnt Signaling. Mol. Med. Rep. 2017, 15, 3840–3846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaspyridonos, M.; McNeill, E.; de Bono, J.P.; Smith, A.; Burnand, K.G.; Channon, K.M.; Greaves, D.R. Galectin-3 Is an Amplifier of Inflammation in Atherosclerotic Plaque Progression through Macrophage Activation and Monocyte Chemoattraction. Arter. Thromb. Vasc. Biol. 2008, 28, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Liu, Z.; Wang, R.; Zheng, Y.; Li, H.; Yang, L. Galectin-3 Is a Potential Mediator for Atherosclerosis. J. Immunol. Res. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, N.C.; Sethi, T. The Regulation of Inflammation by Galectin-3. Immunol. Rev. 2009, 230, 160–171. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-Dependent Phenotypic Modulation of Smooth Muscle Cells Has a Key Role in Atherosclerotic Plaque Pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.P.; Hunziker, P. Atherosclerosis: Insights into Vascular Pathobiology and Outlook to Novel Treatments. J. Cardiovasc. Transl. Res. 2020, 13, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline Biosynthesis Augments Tumor Cell Growth and Aerobic Glycolysis: Involvement of Pyridine Nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Gu, L.; Huang, L.; Fang, J.; Liu, Z.; Xu, Q. The Upregulation of PYCR2 Is Associated with Aggressive Colon Cancer Progression and a Poor Prognosis. Biochem. Biophys. Res. Commun. 2021, 572, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Patriarca, E.J.; Cermola, F.; D’Aniello, C.; Fico, A.; Guardiola, O.; de Cesare, D.; Minchiotti, G. The Multifaceted Roles of Proline in Cell Behavior. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Bedel, A.; Nègre-Salvayre, A.; Heeneman, S.; Grazide, M.-H.; Thiers, J.-C.; Salvayre, R.; Maupas-Schwalm, F. E-Cadherin/β-Catenin/T-Cell Factor Pathway Is Involved in Smooth Muscle Cell Proliferation Elicited by Oxidized Low-Density Lipoprotein. Circ. Res. 2008, 103, 694–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroud, M.J. Linker of Nucleoskeleton and Cytoskeleton Complex Proteins in Cardiomyopathy. Biophys. Rev. 2018, 10, 1033–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson, L.; Otto, H. LUMA Interacts with Emerin and Influences Its Distribution at the Inner Nuclear Membrane. J. Cell Sci. 2008, 121, 536–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroud, M.J.; Banerjee, I.; Veevers, J.; Chen, J. Linker of Nucleoskeleton and Cytoskeleton Complex Proteins in Cardiac Structure, Function, and Disease. Circ. Res. 2014, 114, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.-C.; Mitsuhashi, H.; Keduka, E.; Nonaka, I.; Noguchi, S.; Nishino, I.; Hayashi, Y.K. TMEM43 Mutations in Emery-Dreifuss Muscular Dystrophy-Related Myopathy. Ann. Neurol. 2011, 69, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.; Andersen, C.; Tybjaerg-Hansen, A.; Haunso, S.; Svendsen, J. Mutation Analysis and Evaluation of the Cardiac Localization of TMEM43 in Arrhythmogenic Right Ventricular Cardiomyopathy. Clin. Genet. 2011, 80, 256–264. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.-D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Is a Fully Penetrant, Lethal Arrhythmic Disorder Caused by a Missense Mutation in the TMEM43 Gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Franke, W.W.; Dörflinger, Y.; Kuhn, C.; Zimbelmann, R.; Winter-Simanowski, S.; Frey, N.; Heid, H. Protein LUMA Is a Cytoplasmic Plaque Constituent of Various Epithelial Adherens Junctions and Composite Junctions of Myocardial Intercalated Disks: A Unifying Finding for Cell Biology and Cardiology. Cell Tissue Res. 2014, 357, 159–172. [Google Scholar] [CrossRef]
- Pan, L.; Huang, X.; Liu, Z.-X.; Ye, Y.; Li, R.; Zhang, J.; Wu, G.; Bai, R.; Zhuang, L.; Wei, L.; et al. Inflammatory Cytokine–Regulated TRNA-Derived Fragment TRF-21 Suppresses Pancreatic Ductal Adenocarcinoma Progression. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Vélot, L.; Lessard, F.; Bérubé-Simard, F.-A.; Tav, C.; Neveu, B.; Teyssier, V.; Boudaoud, I.; Dionne, U.; Lavoie, N.; Bilodeau, S.; et al. Proximity-Dependent Mapping of the Androgen Receptor Identifies Kruppel-like Factor 4 as a Functional Partner. Mol. Cell. Proteom. 2021, 20, 100064. [Google Scholar] [CrossRef]
- Szigety, K.M.; Liu, F.; Yuan, C.Y.; Moran, D.J.; Horrell, J.; Gochnauer, H.R.; Cohen, R.N.; Katz, J.P.; Kaestner, K.H.; Seykora, J.T.; et al. HDAC3 Ensures Stepwise Epidermal Stratification via NCoR/SMRT-Reliant Mechanisms Independent of Its Histone Deacetylase Activity. Genes Dev. 2020, 34, 973–988. [Google Scholar] [CrossRef]
- Fearon, I.M.; Gaca, M.D.; Nordskog, B.K. In Vitro Models for Assessing the Potential Cardiovascular Disease Risk Associated with Cigarette Smoking. Toxicol Vitr. 2013, 27, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Giunzioni, I.; Bonomo, A.; Bishop, E.; Castiglioni, S.; Corsini, A.; Bellosta, S. Cigarette Smoke Condensate Affects Monocyte Interaction with Endothelium. Atherosclerosis 2014, 234, 383–390. [Google Scholar] [CrossRef]
- Damiani, I.; Castiglioni, S.; Sochaj-Gregorczyk, A.; Bonacina, F.; Colombo, I.; Rusconi, V.; Otlewski, J.; Corsini, A.; Bellosta, S. Purification and In Vitro Evaluation of an Anti-HER2 Affibody-Monomethyl Auristatin E Conjugate in HER2-Positive Cancer Cells. Biology 2021, 10, 758. [Google Scholar] [CrossRef] [PubMed]
- Bellosta, S.; Selmin, F.; Magri, G.; Castiglioni, S.; Procacci, P.; Sartori, P.; Scarpa, E.; Tolva, V.; Rossi, C.; Puoci, F.; et al. Caffeic Acid-Grafted PLGA as a Novel Material for the Design of Fluvastatin-Eluting Nanoparticles for the Prevention of Neointimal Hyperplasia. Mol. Pharm. 2022, 19, 4333–4344. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Smits, A.H.; van Tilburg, G.B.; Ovaa, H.; Huber, W.; Vermeulen, M. Proteome-Wide Identification of Ubiquitin Interactions Using UbIA-MS. Nat. Protoc. 2018, 13, 530–550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic. Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Bianchi, L.; Gagliardi, A.; Maruelli, S.; Besio, R.; Landi, C.; Gioia, R.; Kozloff, K.M.; Khoury, B.M.; Coucke, P.J.; Symoens, S.; et al. Altered Cytoskeletal Organization Characterized Lethal but Not Surviving Brtl +/− Mice: Insight on Phenotypic Variability in Osteogenesis Imperfecta. Hum. Mol. Genet. 2015, 24, 6118–6133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, L.; Gagliardi, A.; Landi, C.; Focarelli, R.; de Leo, V.; Luddi, A.; Bini, L.; Piomboni, P. Protein Pathways Working in Human Follicular Fluid: The Future for Tailored IVF? Expert. Rev. Mol. Med. 2016, 18, e9. [Google Scholar] [CrossRef]
- Gagliardi, A.; Besio, R.; Carnemolla, C.; Landi, C.; Armini, A.; Aglan, M.; Otaify, G.; Temtamy, S.A.; Forlino, A.; Bini, L.; et al. Cytoskeleton and Nuclear Lamina Affection in Recessive Osteogenesis Imperfecta: A Functional Proteomics Perspective. J. Proteom. 2017, 167, 46–59. [Google Scholar] [CrossRef]
- Bianchi, L.; Altera, A.; Barone, V.; Bonente, D.; Bacci, T.; de Benedetto, E.; Bini, L.; Tosi, G.M.; Galvagni, F.; Bertelli, E. Untangling the Extracellular Matrix of Idiopathic Epiretinal Membrane: A Path Winding among Structure, Interactomics and Translational Medicine. Cells 2022, 11, 2531. [Google Scholar] [CrossRef] [PubMed]
- Vantaggiato, L.; Shaba, E.; Carleo, A.; Bezzini, D.; Pannuzzo, G.; Luddi, A.; Piomboni, P.; Bini, L.; Bianchi, L. Neurodegenerative Disorder Risk in Krabbe Disease Carriers. Int. J. Mol. Sci. 2022, 23, 13537. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProtKB Protein Name | UniProtKB A.N. | Gene Symbol (Alternative Symbol) | CSC Abundance | Control Abundance | Coverage (Unique Peptides) | Score | Adj. p Value |
|---|---|---|---|---|---|---|---|
| Atrial natriuretic peptide receptor 3 | P17342 | NPR3 (ANPRC, C5orf23, NPRC) | 0 (0) | 22.8 (0.2) | 28.1% (9) | 22.7 | 8.08 × 10−4 |
| Collectin-12 | Q5KU26 | COLEC12 (CLP1, NSR2, SCARA4, SRCL) | 0 (0) | 21.6 (0.2) | 7.7% (6) | 8.2 | 7.92 × 10−4 |
| Elongator complex protein 2 | Q6IA86 | ELP2 (STATIP1) | 7.1 (0.0) | 21.6 (0.0) | 9.2% (5) | 18.4 | 5.28 × 10−4 |
| (E3-independent) E2 ubiquitin-conjugating enzyme | Q9C0C9 | UBE2O (KIAA1734) | 7.3 (0.0) | 22.1 (0.2) | 7.3% (6) | 20.5 | 1.17 × 10−3 |
| Interferon-induced, double-stranded RNA-activated protein kinase | P19525 | EIF2AK2 (PKR, PRKR) | 7.4 (0.0) | 0 (0) | 14.8% (7) | 13.2 | 9.32 × 10−4 |
| Protein MON2 homolog | Q7Z3U7 | MON2 (KIAA1040, SF21) | 0 (0) | 20.2 (0.0) | 2% (3) | 5.0 | 5.04 × 10−4 |
| Pyrroline-5-carboxylate reductase 2 | Q96C36 | PYCR2 | 14 (0.1) | 0 (0) | 15.9% (2) | 5.0 | 8.45 × 10−4 |
| Schlafen family member 5 | Q08AF3 | SLFN5 | 7 (0.0) | 21.2 (0.2) | 8% (5) | 11.3 | 1.34 × 10−3 |
| Serine/arginine-rich splicing factor 5 | Q13243 | SRSF5 (HRS, SFRS5, SRP40) | 7.4 (0.1) | 22.3 (0.2) | 11.4% (2) | 4.4 | 1.19 × 10−3 |
| Transmembrane emp24 domain-containing protein 1 | Q13445 | TMED1 (IL1RL1L, IL1RL1LG, ST2L) | 7.3 (0.0) | 22.1 (0.1) | 9.7% (2) | 4.0 | 5.28 × 10−4 |
| Transmembrane protein 43 | Q9BTV4 | TMEM43 | 7.3 (0.1) | 22.1 (0.4) | 22% (6) | 20.2 | 4.64 × 10−3 |
| Gene Name | Sequences | Gene Name | Sequences |
|---|---|---|---|
| Abca1 | FW 5′-AAAACCGCAGACATCCTTCAG-3′ RV 5′-CATACCGAAACTCGTTCACCC-3′ | Klf4 | FW 5′-CTTTCCTGCCAGACCAGATG-3′ RV 5′-GGTTTCTCGCCTGTGTGAGT-3′ |
| Abcg1 | FW 5′-CCTTATCAATGGAATGCCCCG-3′ RV 5′-CTGCCTTCATCCTTCTCCTG-3′ | Lgals-3 | FW 5′-TGGGCACAGTGAAACCCAAC-3′ RV 5′-TCCTGCTTCGTGTTACACACA-3′ |
| Acta2 | FW 5′-GTCCCAGACATCAGGGAGTAA-3′ RV 5′-TCGGATACTTCAGCGTCAGGA-3′ | Myocd | FW 5′-AAGGTCCATTCCAACTGCTC-3′ RV 5′-CCATCTCTACTGCTGTCATCC-3′ |
| Cnn1 | FW 5′-TTGAGAGAAGGCAGGAACATC-3′ RV 5′-GTACCCAGTTTGGGATCATAGAG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchi, L.; Damiani, I.; Castiglioni, S.; Carleo, A.; De Salvo, R.; Rossi, C.; Corsini, A.; Bellosta, S. Smooth Muscle Cell Phenotypic Switch Induced by Traditional Cigarette Smoke Condensate: A Holistic Overview. Int. J. Mol. Sci. 2023, 24, 6431. https://doi.org/10.3390/ijms24076431
Bianchi L, Damiani I, Castiglioni S, Carleo A, De Salvo R, Rossi C, Corsini A, Bellosta S. Smooth Muscle Cell Phenotypic Switch Induced by Traditional Cigarette Smoke Condensate: A Holistic Overview. International Journal of Molecular Sciences. 2023; 24(7):6431. https://doi.org/10.3390/ijms24076431
Chicago/Turabian StyleBianchi, Laura, Isabella Damiani, Silvia Castiglioni, Alfonso Carleo, Rossana De Salvo, Clara Rossi, Alberto Corsini, and Stefano Bellosta. 2023. "Smooth Muscle Cell Phenotypic Switch Induced by Traditional Cigarette Smoke Condensate: A Holistic Overview" International Journal of Molecular Sciences 24, no. 7: 6431. https://doi.org/10.3390/ijms24076431
APA StyleBianchi, L., Damiani, I., Castiglioni, S., Carleo, A., De Salvo, R., Rossi, C., Corsini, A., & Bellosta, S. (2023). Smooth Muscle Cell Phenotypic Switch Induced by Traditional Cigarette Smoke Condensate: A Holistic Overview. International Journal of Molecular Sciences, 24(7), 6431. https://doi.org/10.3390/ijms24076431