
BioGraph: Data Model for Linking and Querying Diverse Biological Metadata



Abstract
:1. Introduction
Related Work
2. Results and Discussion
3. Methods and Materials
3.1. Model Definition and Implementation
- Unified representation of metadata from diverse formats and schemas.
- Efficient querying and finding complex patterns in data.
- Allowing easy extensibility through schema extensions without affecting the core data model.
- Being database-agnostic as much as possible, so the underlying database management system and related software do not dictate the model’s design.
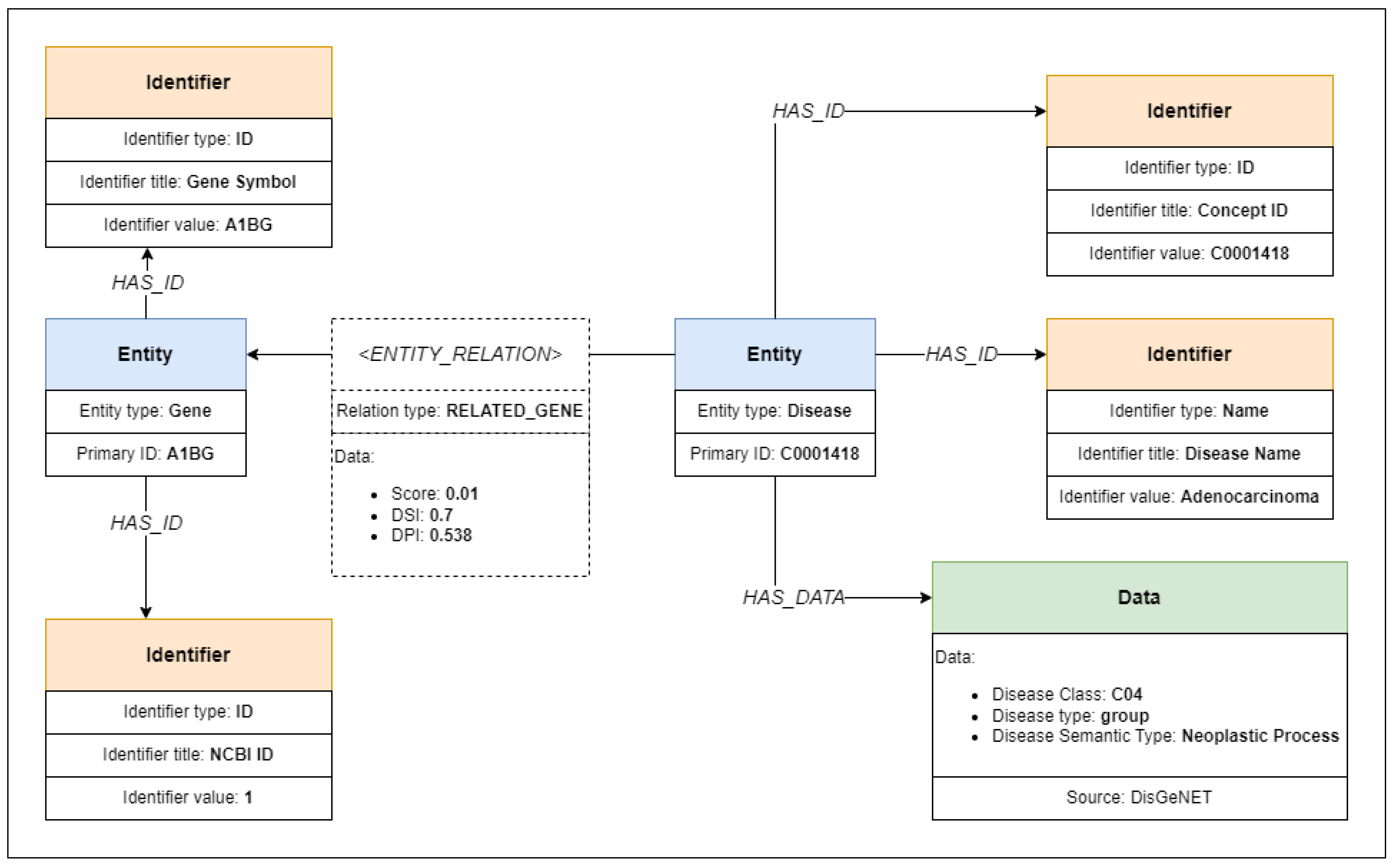
3.2. Model Definition
3.3. Model Implementation
3.4. Data Flows
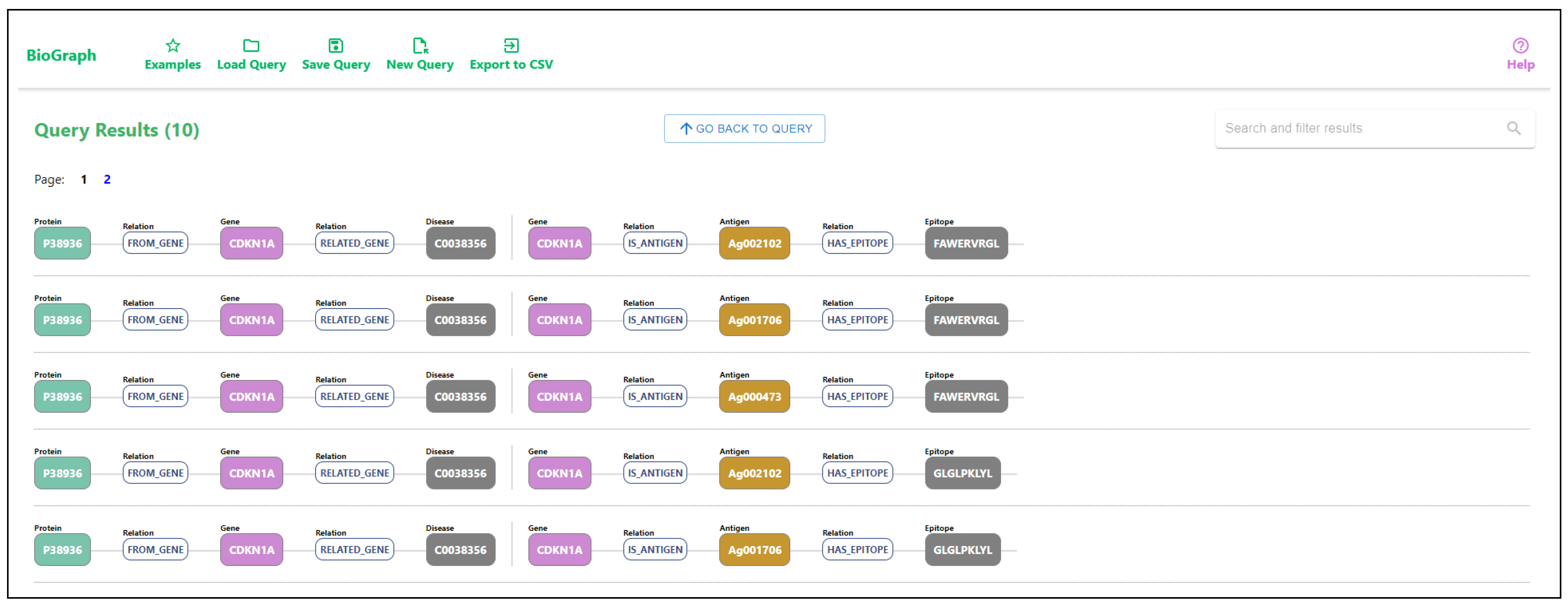
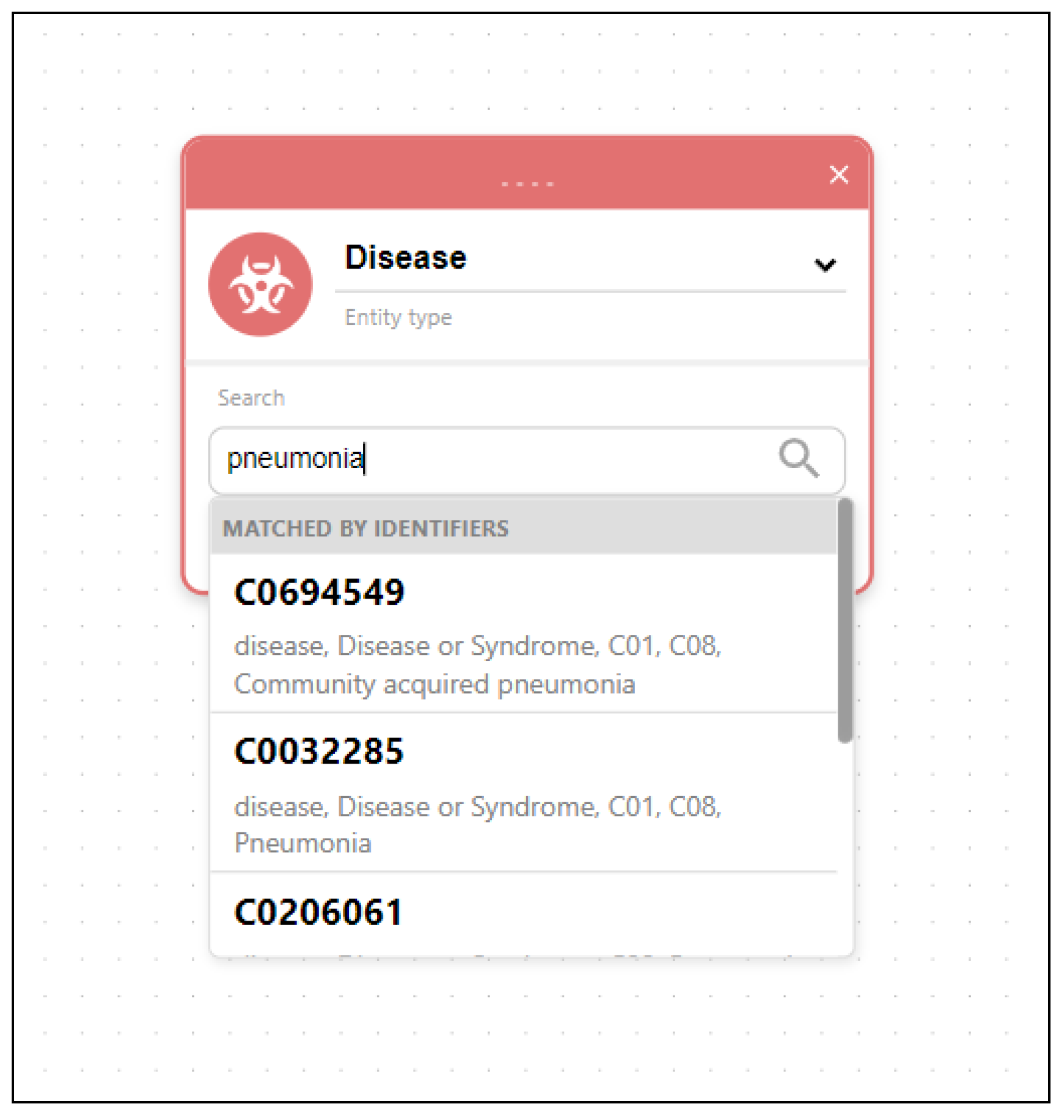
3.5. Searching the Data
3.6. Materials
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| API | Application Programming Interface |
| CSV | Comma-Separated Values |
| DisGeNET | Disease Gene Network |
| DisProt | Database of Protein Disorder |
| HGNC | HUGO Gene Nomenclature Committee |
| HUGO | Human Genome Organization |
| IEDB | Immune Epitope DataBase |
| JSON | JavaScript Object Notation |
| KG | Knowledge Graph |
| NCBI | National Center for Biotechnology Information |
| OBO | Open Biomedical Ontologies |
| RDF | Resource Description Framework |
| ROBOKOP | Reasoning Over Biomedical Objects linked in Knowledge Oriented Pathways |
| TSV | Tab-Separated Values |
| UniProt | Universal Protein Resource |
References
- Piovesan, D.; Del Conte, A.; Clementel, D.; Monzon, A.M.; Bevilacqua, M.; Aspromonte, M.C.; Iserte, J.A.; Orti, F.E.; Marino-Buslje, C.; Tosatto, S.C. MobiDB: 10 years of intrinsically disordered proteins. Nucleic Acids Res. 2023, 51, D438–D444. [Google Scholar] [CrossRef]
- Azeem, M.; Jamil, M.K.; Shang, Y. Notes on the localization of generalized hexagonal cellular networks. Mathematics 2023, 11, 844. [Google Scholar] [CrossRef]
- Azeem, M.; Jamil, M.K.; Javed, A. Verification of some topological indices of Y-junction based nanostructures by M-polynomials. J. Math. 2022, 2022, 8238651. [Google Scholar] [CrossRef]
- Koam, A.N.; Ahmad, A.; Husain, S.; Azeem, M. Mixed metric dimension of hollow coronoid structure. Ain Shams Eng. J. 2023, 14, 102000. [Google Scholar] [CrossRef]
- Liu, J.; Lei, X.; Zhang, Y.; Pan, Y. The prediction of molecular toxicity based on BiGRU and GraphSAGE. Comput. Biol. Med. 2023, 153, 106524. [Google Scholar] [CrossRef] [PubMed]
- Veličković, P. Everything is connected: Graph neural networks. Curr. Opin. Struct. Biol. 2023, 79, 102538. [Google Scholar] [CrossRef] [PubMed]
- Chandak, P.; Huang, K.; Zitnik, M. Building a knowledge graph to enable precision medicine. Sci. Data 2023, 10, 67. [Google Scholar] [CrossRef]
- McBride, B. The resource description framework (RDF) and its vocabulary description language RDFS. In Handbook on Ontologies; Springer: Berlin/Heidelberg, Germany, 2004; pp. 51–65. [Google Scholar] [CrossRef]
- Angles, R.; Thakkar, H.; Tomaszuk, D. RDF and Property Graphs Interoperability: Status and Issues. AMW 2019, 2369, 1–11. [Google Scholar]
- Smith, B.; Ashburner, M.; Rosse, C.; Bard, J.; Bug, W.; Ceusters, W.; Goldberg, L.J.; Eilbeck, K.; Ireland, A.; Mungall, C.J.; et al. The OBO Foundry: Coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007, 25, 1251–1255. [Google Scholar] [CrossRef] [Green Version]
- Unni, D.R.; Moxon, S.A.; Bada, M.; Brush, M.; Bruskiewich, R.; Caufield, J.H.; Clemons, P.A.; Dancik, V.; Dumontier, M.; Fecho, K.; et al. Biolink Model: A universal schema for knowledge graphs in clinical, biomedical, and translational science. Clin. Transl. Sci. 2022, 15, 1848–1855. [Google Scholar] [CrossRef]
- Bizon, C.; Cox, S.; Balhoff, J.; Kebede, Y.; Wang, P.; Morton, K.; Fecho, K.; Tropsha, A. ROBOKOP KG and KGB: Integrated Knowledge Graphs from Federated Sources. J. Chem. Inf. Model. 2019, 59, 4968–4973. [Google Scholar] [CrossRef] [PubMed]
- Shefchek, K.A.; Harris, N.L.; Gargano, M.; Matentzoglu, N.; Unni, D.; Brush, M.; Keith, D.; Conlin, T.; Vasilevsky, N.; Zhang, X.A.; et al. The Monarch Initiative in 2019: An integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 2020, 48, D704–D715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koza. Available online: https://koza.monarchinitiative.org/ (accessed on 28 December 2022).
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef]
- Elsevier. Biology Knowledge Graph. Available online: https://www.elsevier.com/solutions/biology-knowledge-graph (accessed on 28 December 2022).
- Ji, S.; Pan, S.; Cambria, E.; Marttinen, P.; Yu, P.S. A Survey on Knowledge Graphs: Representation, Acquisition, and Applications. IEEE Trans. Neural Netw. Learn. Syst. 2022, 33, 494–514. [Google Scholar] [CrossRef]
- Wu, X.; Duan, J.; Pan, Y.; Li, M. Medical knowledge graph: Data sources, construction, reasoning, and applications. Big Data Min. Anal. 2023, 6, 201–217. [Google Scholar] [CrossRef]
- Vucetic, S.; Obradovic, Z.; Vacic, V.; Radivojac, P.; Peng, K.; Iakoucheva, L.M.; Cortese, M.S.; Lawson, J.D.; Brown, C.J.; Sikes, J.G.; et al. DisProt: A database of protein disorder. Bioinformatics 2004, 21, 137–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piñero, J.; Juan Manuel, R.A.; Josep Saüch-Pitarch, F.R.; Emilio Centeno, F.S.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucl. Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Chitkushev, L.; Olsen, L.R.; Keskin, D.B.; Brusic, V. TANTIGEN 2.0: A knowledge base of tumor T cell antigens and epitopes. BMC Bioinform. 2021, 22, 40. [Google Scholar] [CrossRef]
- IEDB. Available online: http://www.iedg.org (accessed on 6 February 2023).
- Seal, R.L.; Braschi, B.; Gray, K.; Jones, T.E.; Tweedie, S.; Haim-Vilmovsky, L.; Bruford, E.A. Genenames. org: The HGNC resources in 2023. Nucleic Acids Res. 2023, 51, D1003–D1009. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R.; et al. Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Neo4j Graph Database. Available online: https://neo4j.com/product/neo4j-graph-database/ (accessed on 6 February 2023).
- NodeJS. Available online: https://nodejs.org/ (accessed on 6 February 2023).
- Pezoa, F.; Reutter, J.L.; Suarez, F.; Ugarte, M.; Vrgoč, D. Foundations of JSON schema. In Proceedings of the 25th International Conference on World Wide Web, Montréal, QC, Canada, 11–15 April 2016; International World Wide Web Conferences Steering Committee: Geneva, Switzerland, 2016; pp. 263–273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Li, S.; Chen, M. Characterization and Function of Circular RNAs in Plants. Front. Mol. Biosci. 2020, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Wang, J.; Harrison, A.P.; Meng, X.; Chen, D.; Chen, M. Genome-wide view of natural antisense transcripts in Arabidopsis thaliana. DNA Res. 2015, 22, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Ivanisenko, T.V.; Demenkov, P.S.; Kolchanov, N.A.; Ivanisenko, V.A. The New Version of the ANDDigest Tool with Improved AI-Based Short Names Recognition. Int. J. Mol. Sci. 2022, 23, 14934. [Google Scholar] [CrossRef] [PubMed]
- Ivanisenko, T.V.; Saik, O.V.; Demenkov, P.S.; Ivanisenko, N.V.; Savostianov, A.N.; Ivanisenko, V.A. ANDDigest: A new web-based module of ANDSystem for the search of knowledge in the scientific literature. BMC Bioinform. 2020, 21, 228. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criterium | BioGraph | ROBOKOP | Monarch | GeneCards | Elsevier Biology KG |
|---|---|---|---|---|---|
| Open-source | ✓ | ✓ | ✓ | ✗ | ✗ |
| Local deployment | ✓ | ✓ | ✓ | ✗ | ✗ |
| Pattern querying | ✓ | ✓ | ✗ | ✗ | ? |
| Graphical queries | ✓ | ✓ | ✗ | ✗ | ? |
| Extensibility | ✓ | ✗ | ✗ | ? | ? |
| Natural Language Queries | ✗ | ✓ | ✗ | ✗ | ? |
| Loading and Storing Queries | ✓ | ✓ | ✗ | ✗ | ? |
| User-friendly | ✓ | ✗ | ✓ | ✓ | ? |
| Relation | Description | Example |
|---|---|---|
| IS | Relation representing equality between objects, where object A, on one side of the relation can also be represented as B in general or specific circumstances. The relation can contain details specifying the equality relation. | Gene A IS Antigen B |
| IS INSTANCE | Relation between objects where one of the objects is an instance of a bigger class. | TP53 protein in humans IS INSTANCE of TP53 protein. |
| IS VARIANT | Representing relation between objects where one object is an isoform of the other. | Protein A IS VARIANT of protein B |
| FROM | Represents the relation between the object and its source. | Protein A FROM gene B. Gene C FROM organism D |
| CONTAINS | Represents the relation between the object and its part. | Antigen A CONTAINS Epitope B |
| RELATED WITH | General relationship between objects. A weight, or relation score, of the relation can be defined in relation parameters. | Gene A is RELATED WITH disease B, with a relation score of 0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veljković, A.N.; Orlov, Y.L.; Mitić, N.S. BioGraph: Data Model for Linking and Querying Diverse Biological Metadata. Int. J. Mol. Sci. 2023, 24, 6954. https://doi.org/10.3390/ijms24086954
Veljković AN, Orlov YL, Mitić NS. BioGraph: Data Model for Linking and Querying Diverse Biological Metadata. International Journal of Molecular Sciences. 2023; 24(8):6954. https://doi.org/10.3390/ijms24086954
Chicago/Turabian StyleVeljković, Aleksandar N., Yuriy L. Orlov, and Nenad S. Mitić. 2023. "BioGraph: Data Model for Linking and Querying Diverse Biological Metadata" International Journal of Molecular Sciences 24, no. 8: 6954. https://doi.org/10.3390/ijms24086954
APA StyleVeljković, A. N., Orlov, Y. L., & Mitić, N. S. (2023). BioGraph: Data Model for Linking and Querying Diverse Biological Metadata. International Journal of Molecular Sciences, 24(8), 6954. https://doi.org/10.3390/ijms24086954