
Investigation of the Compatibility between Warheads and Peptidomimetic Sequences of Protease Inhibitors—A Comprehensive Reactivity and Selectivity Study

 , , , , , , , , ,
, , , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
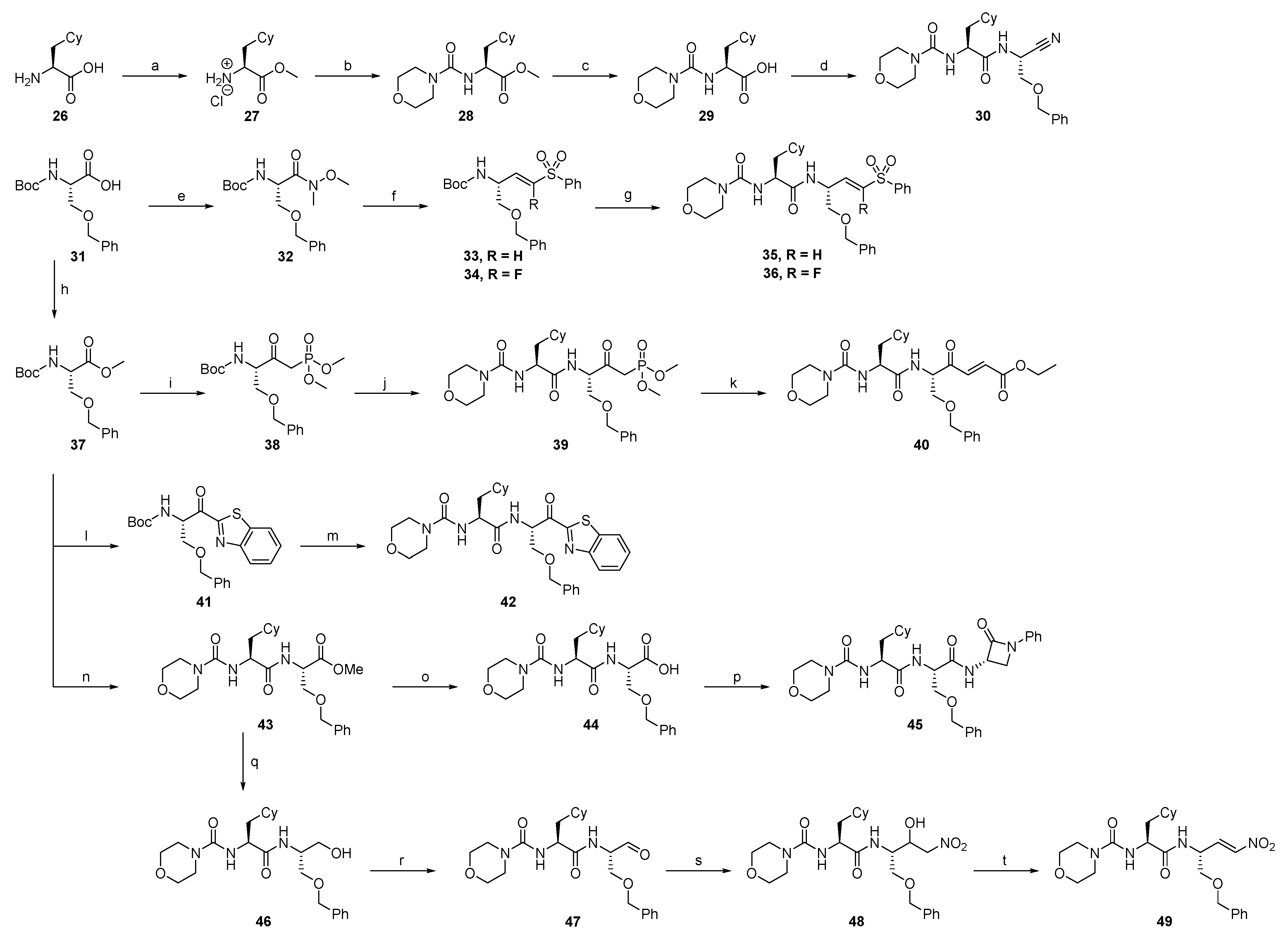{kind=link}
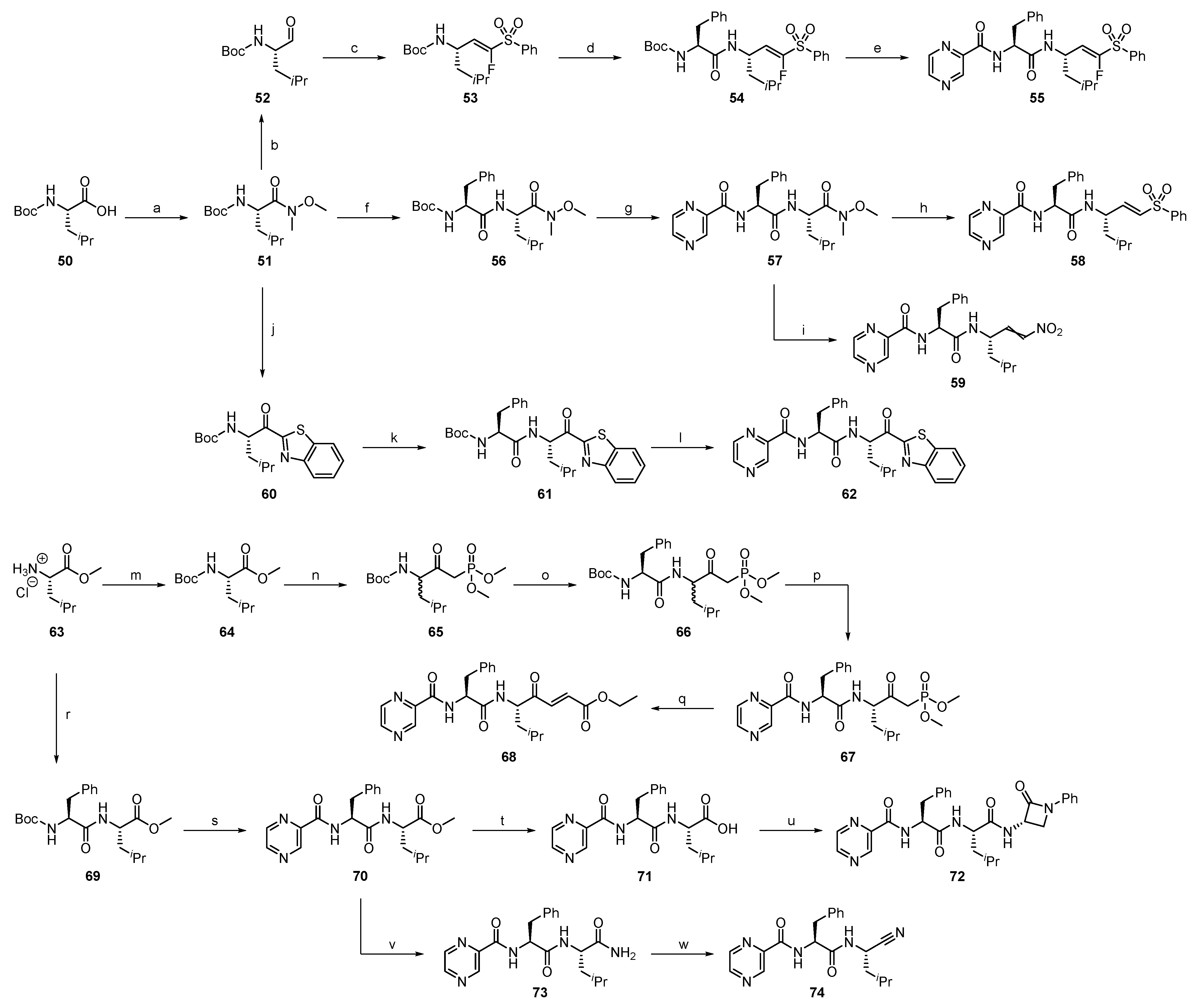{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
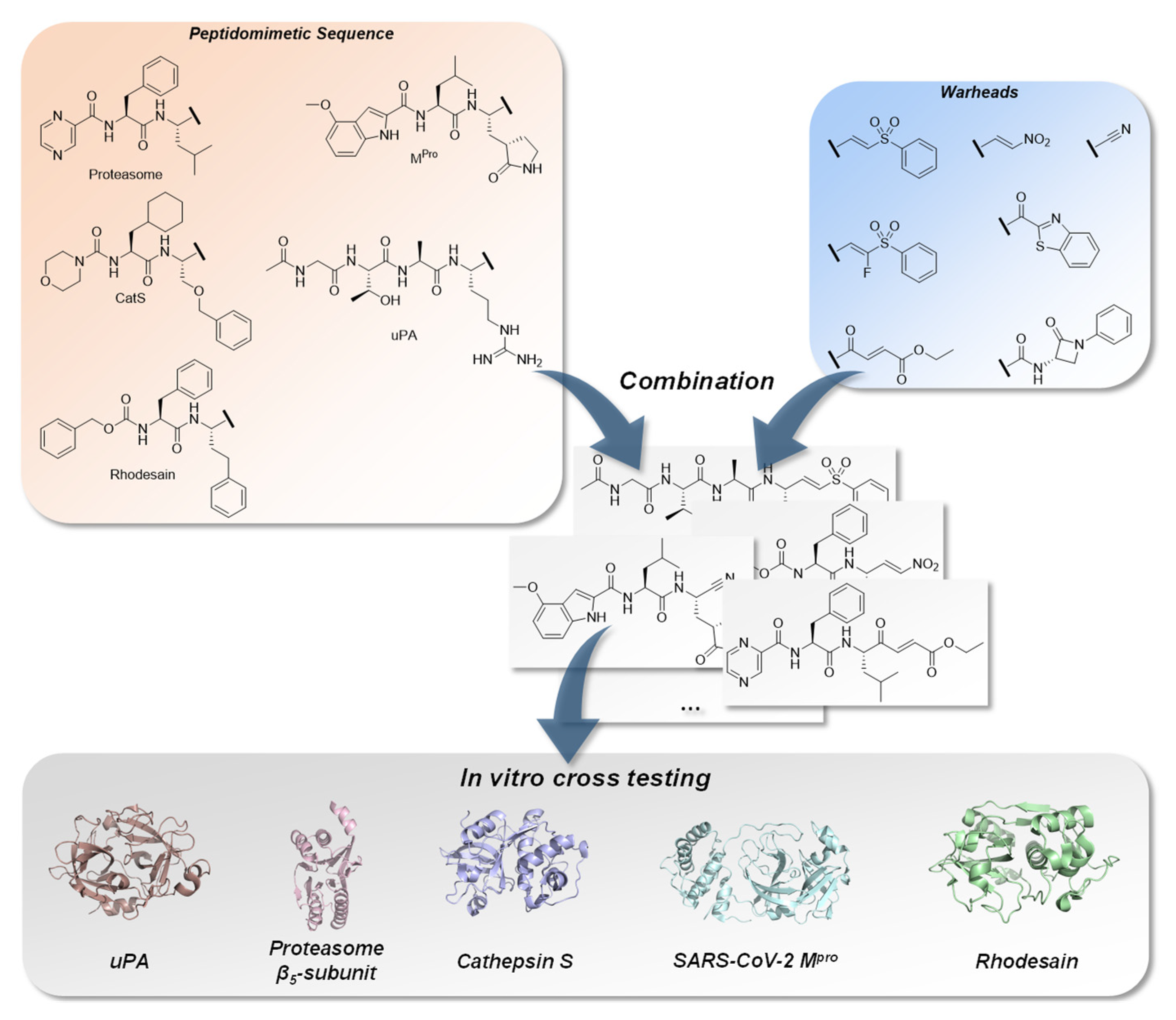
:1. Introduction

2. Results
2.1. Chemistry
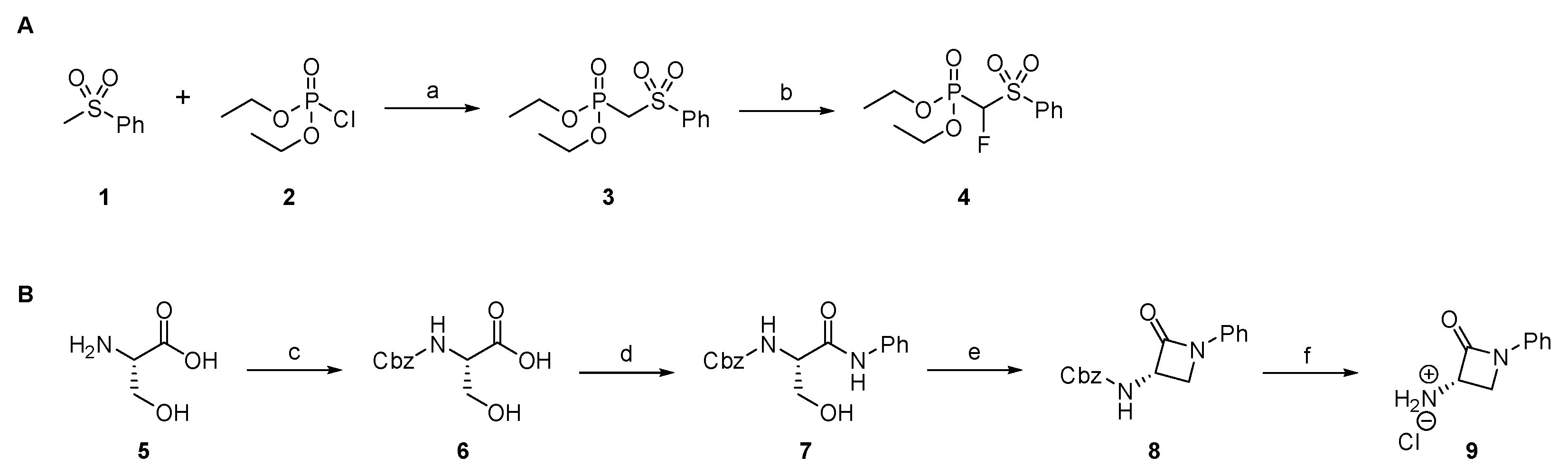
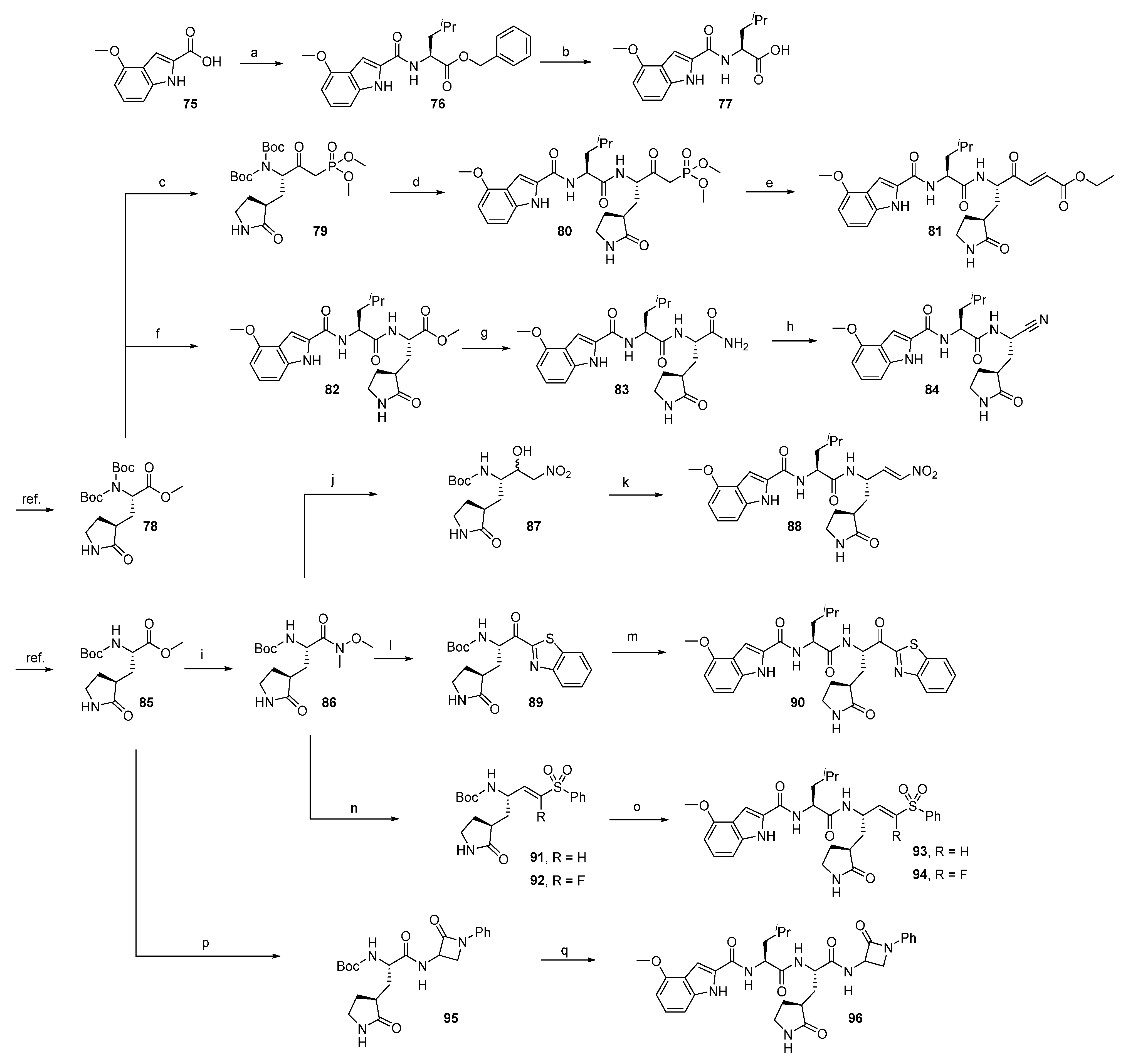
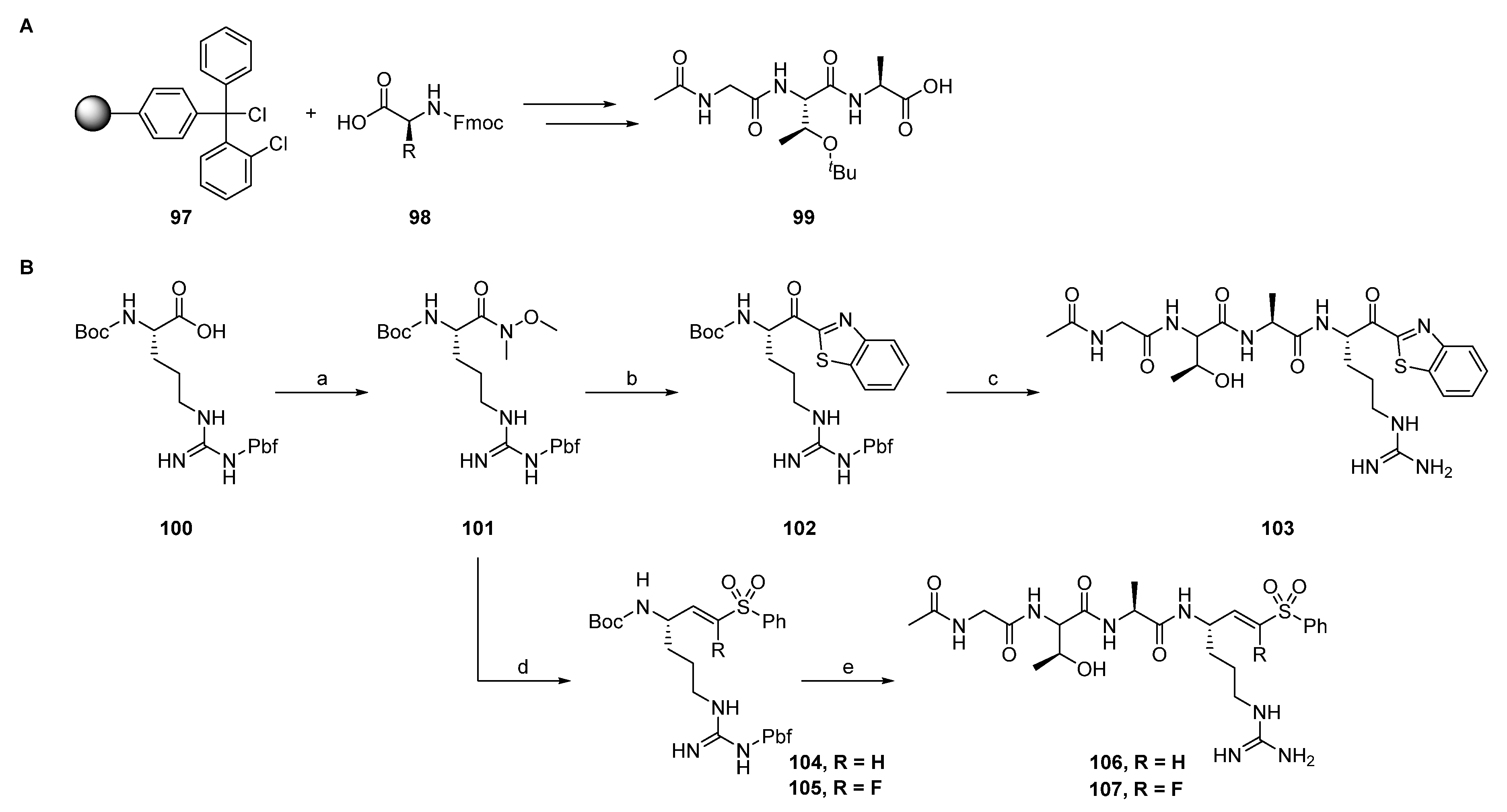
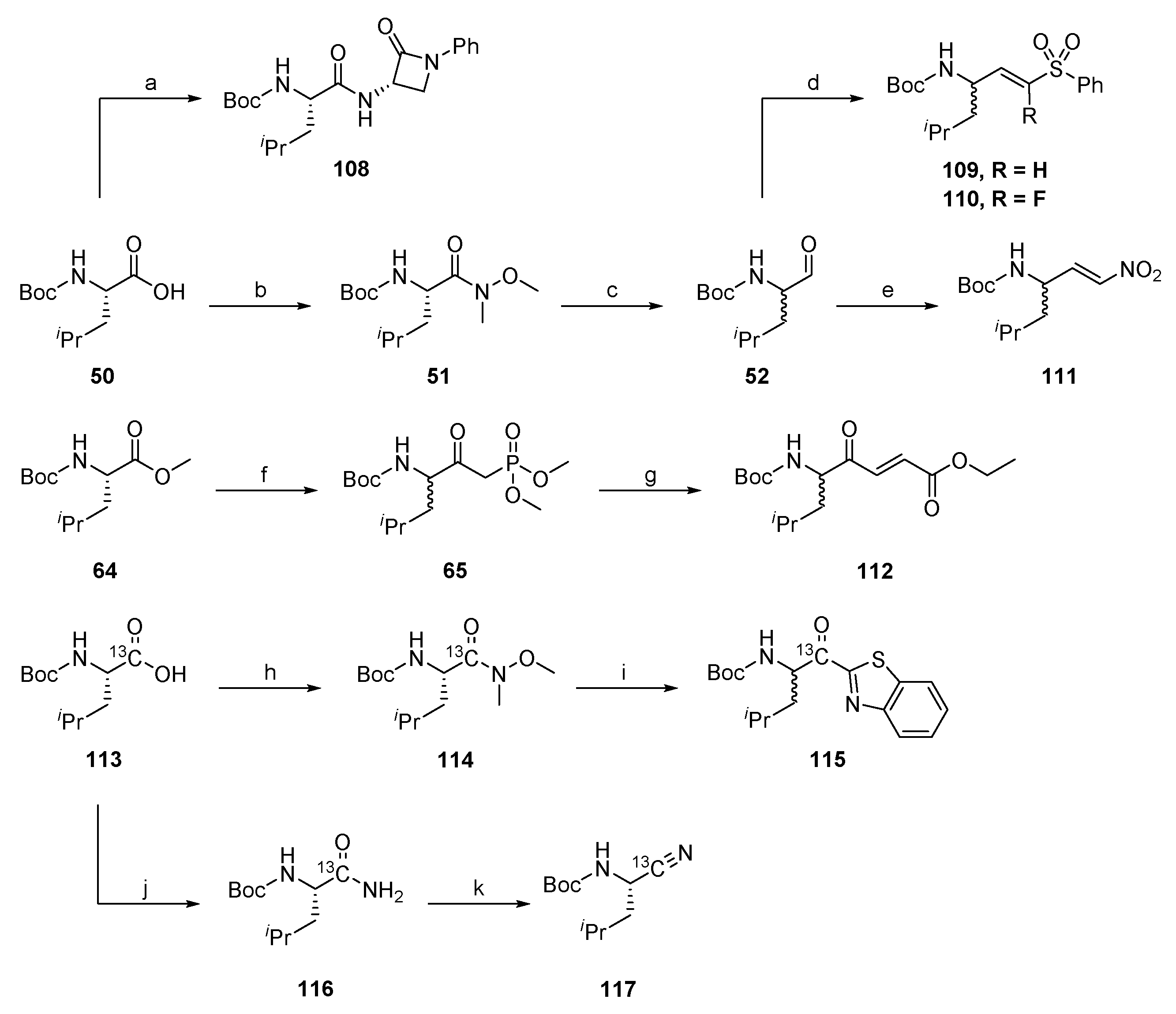
2.1.1. Synthesis of Precursors
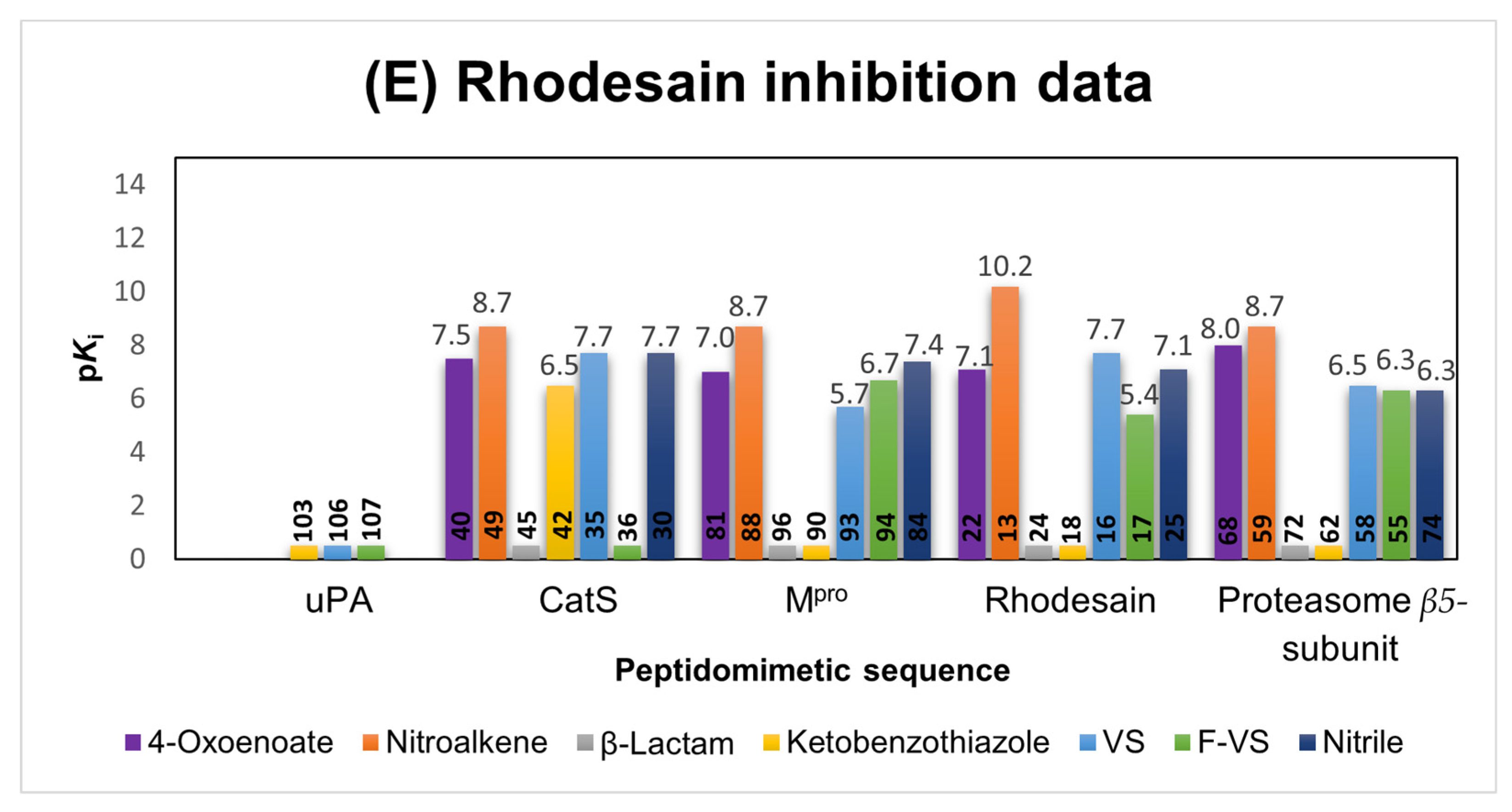
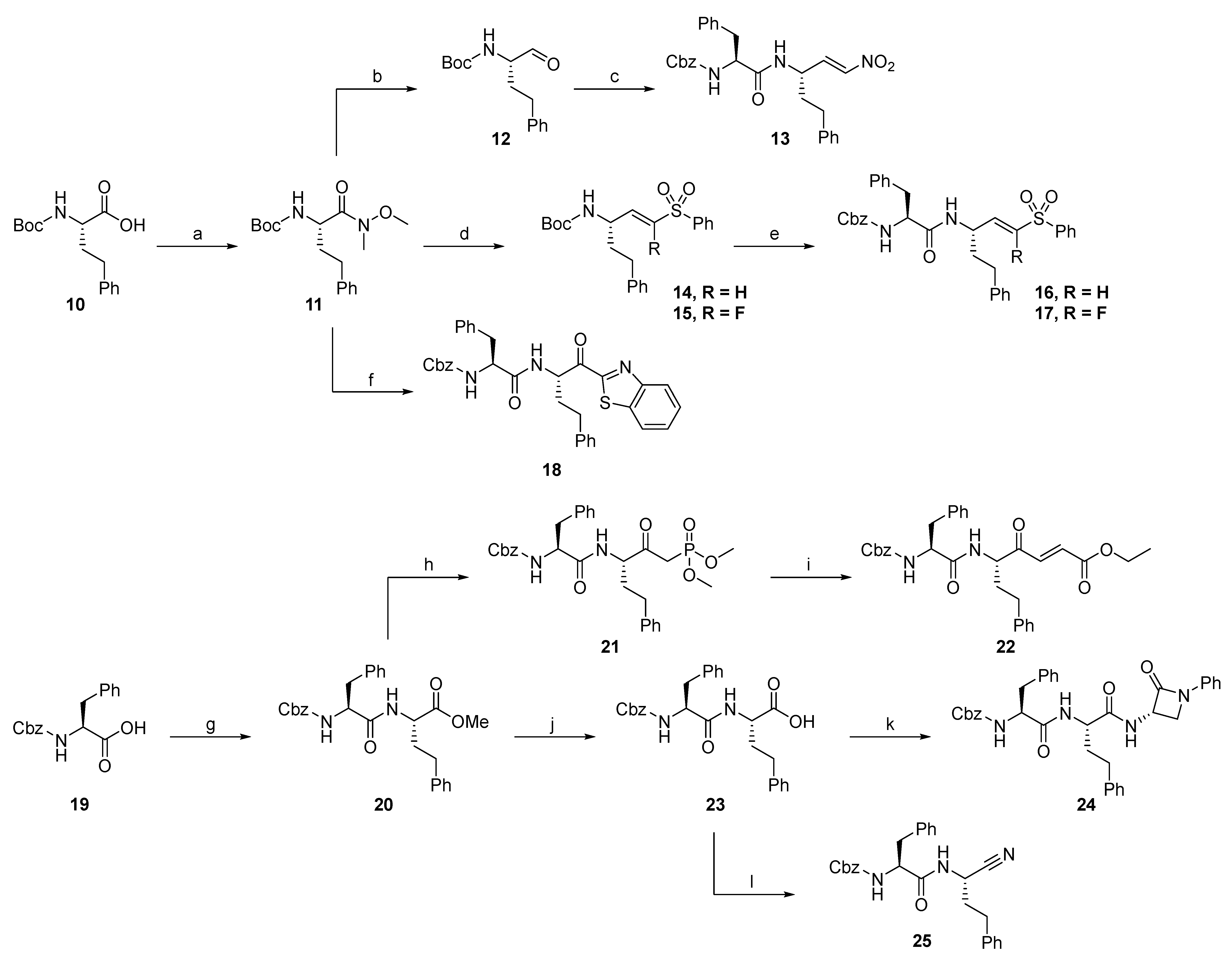
2.1.2. Rhodesain Inhibitors
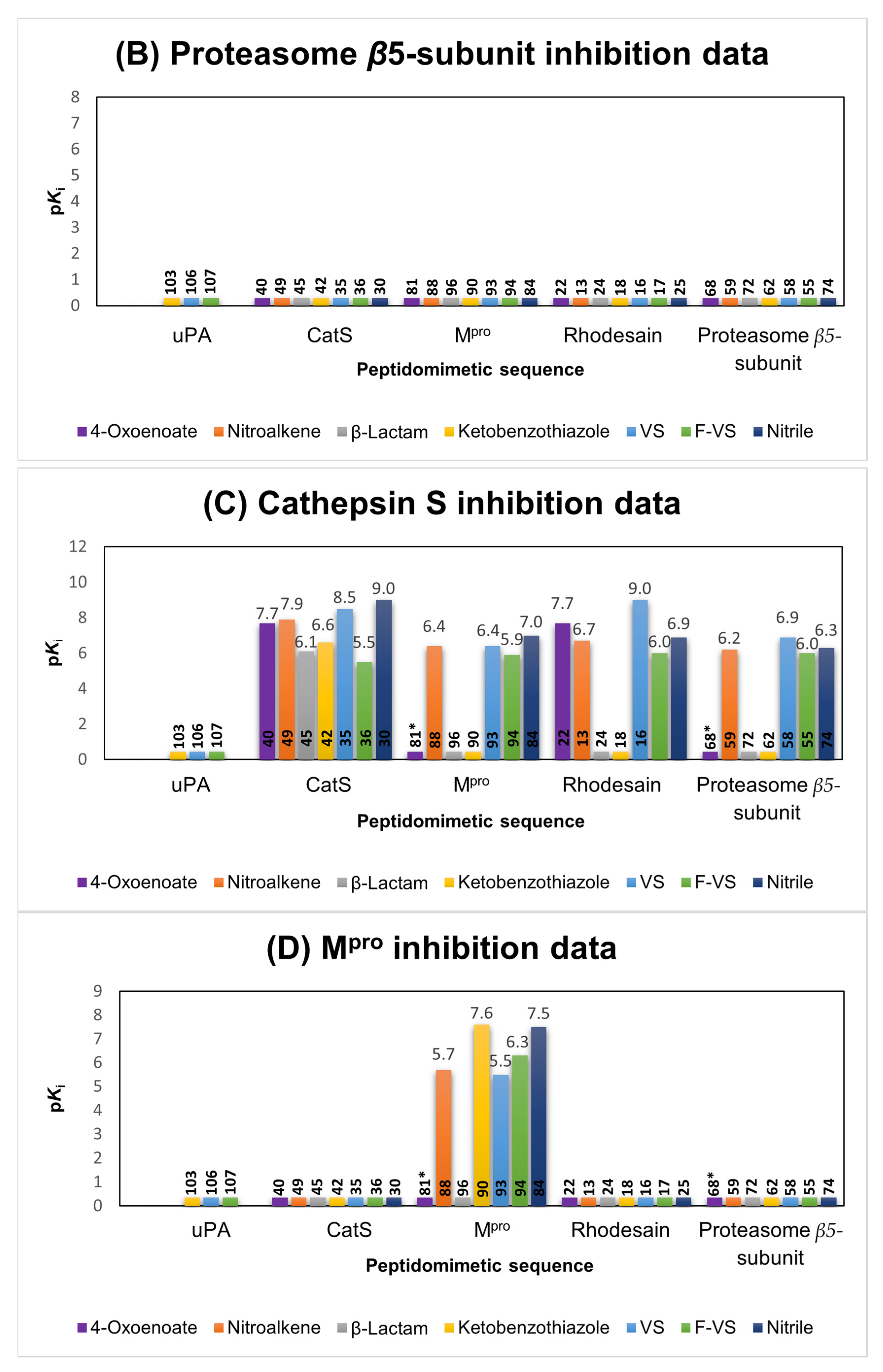
2.1.3. Cathepsin S Inhibitors
2.1.4. Proteasome β5-Subunit Inhibitors
2.1.5. SARS-CoV-2 Mpro Inhibitors
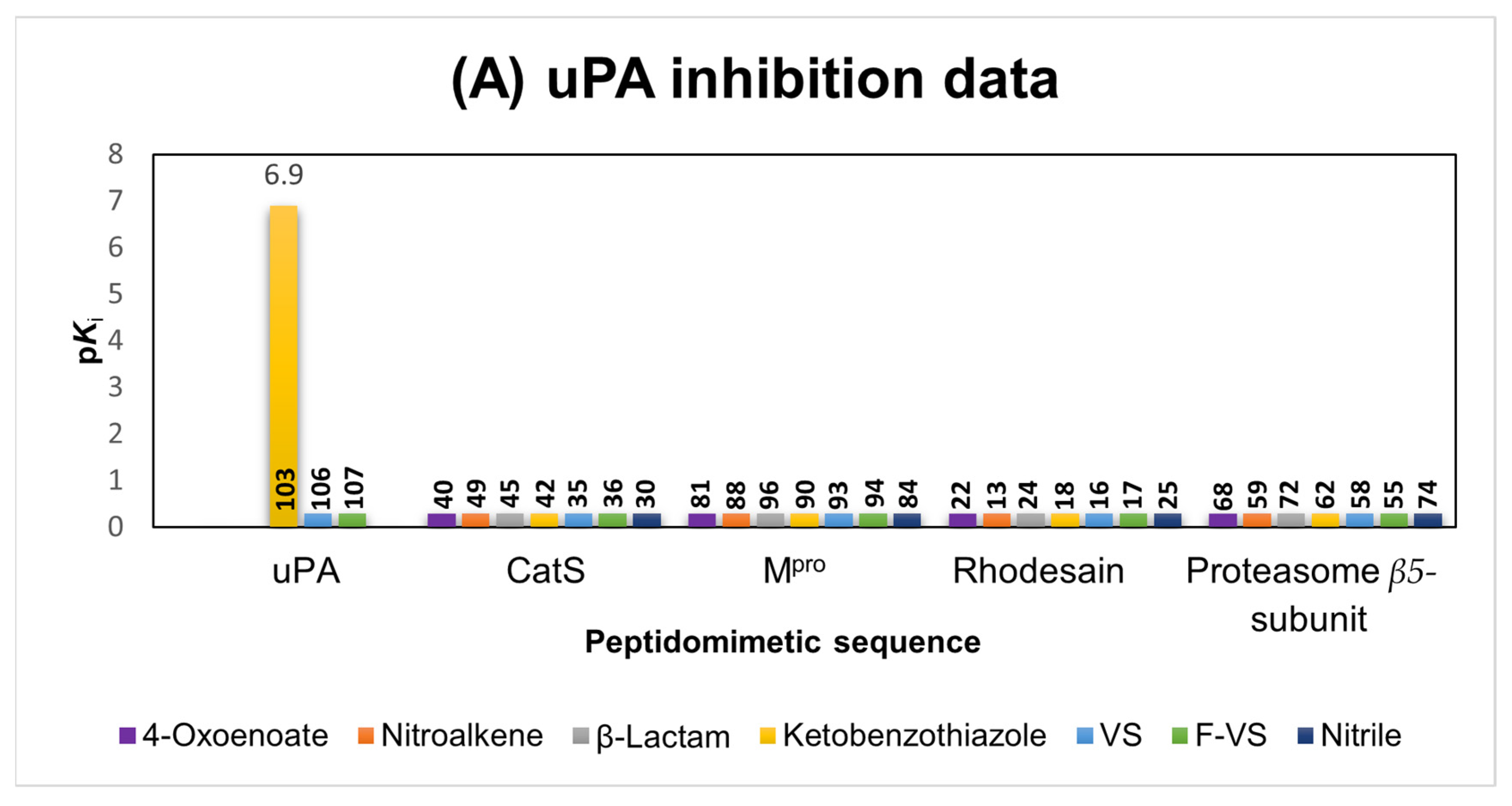
2.1.6. uPA Inhibitors
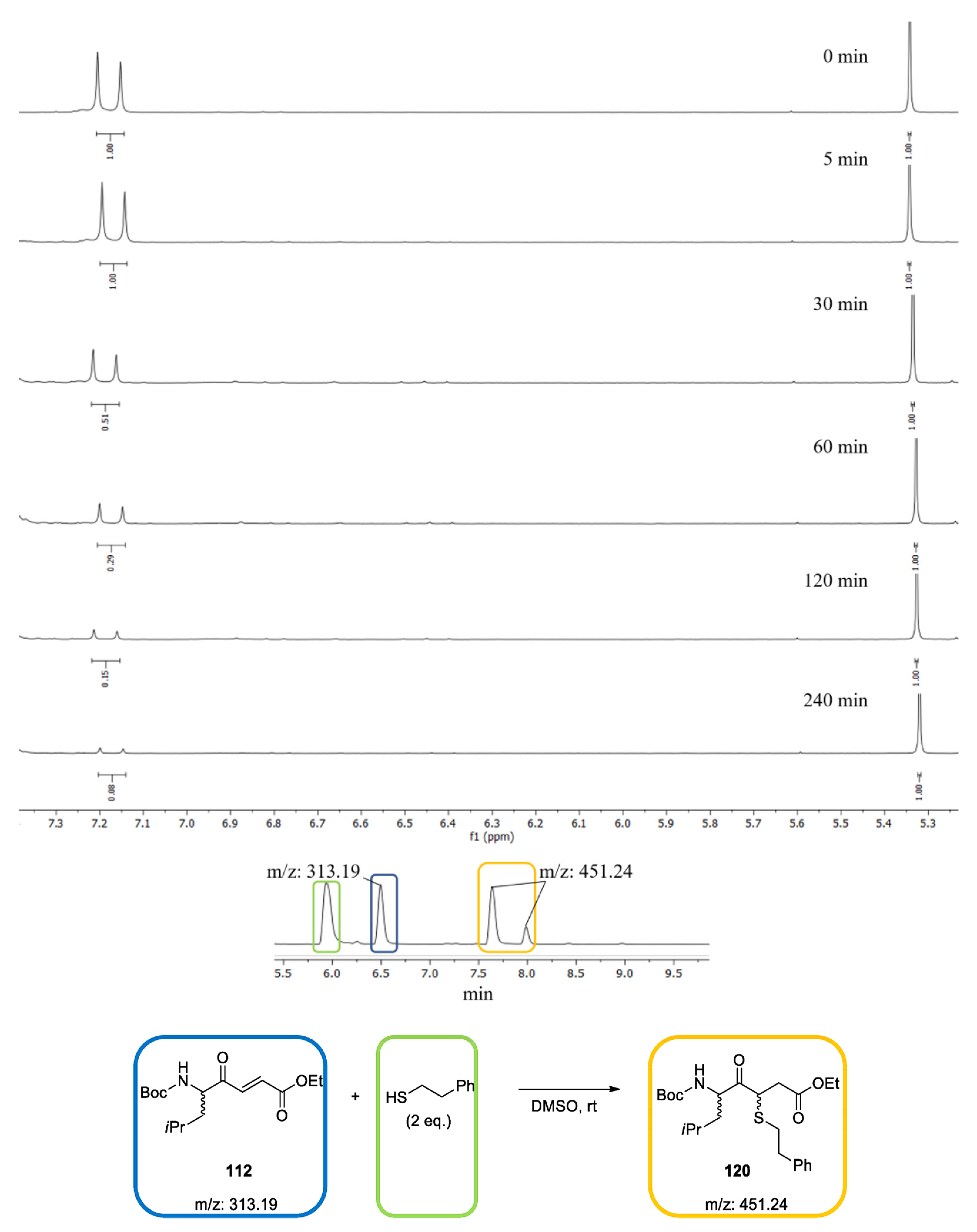
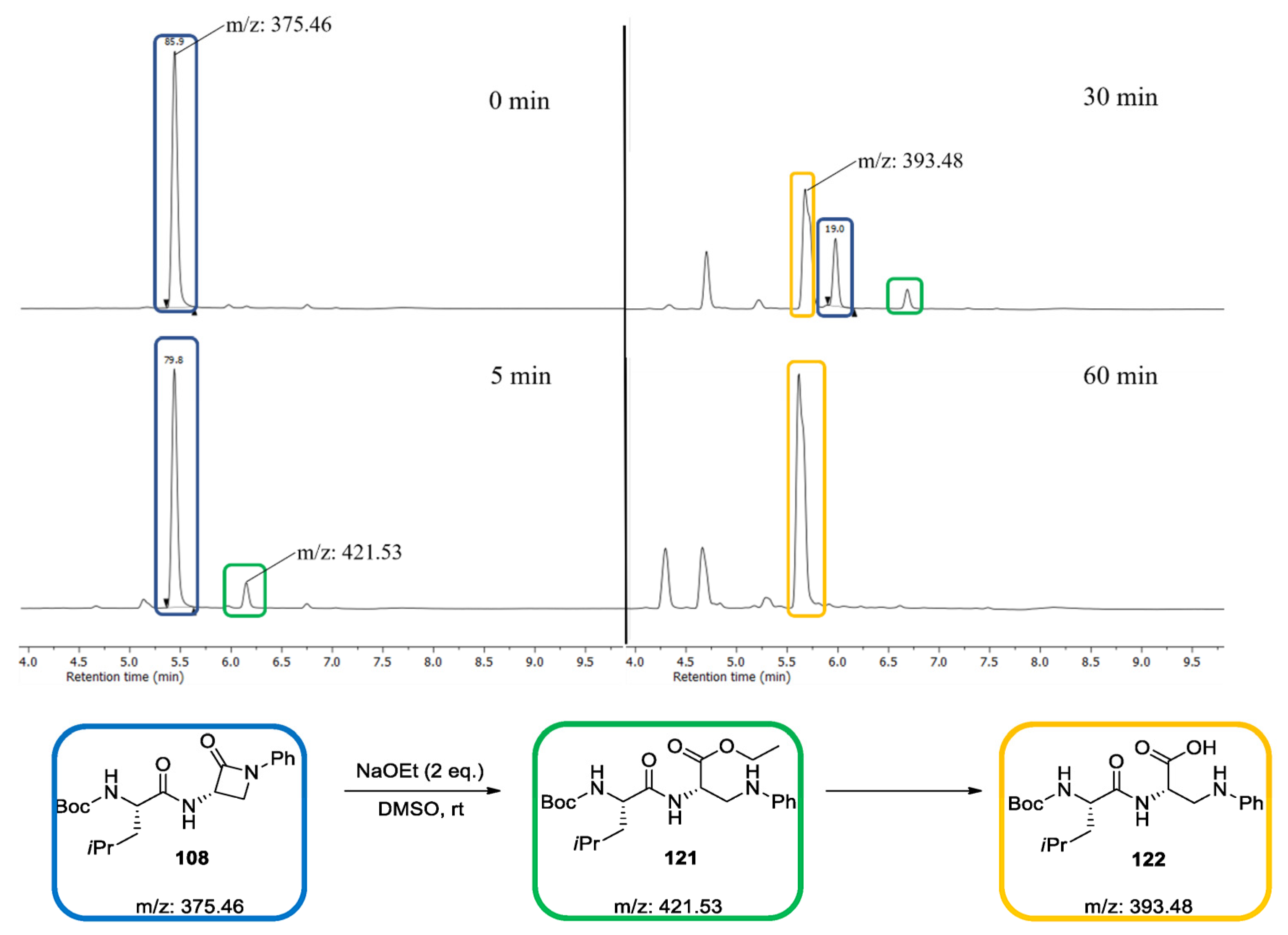
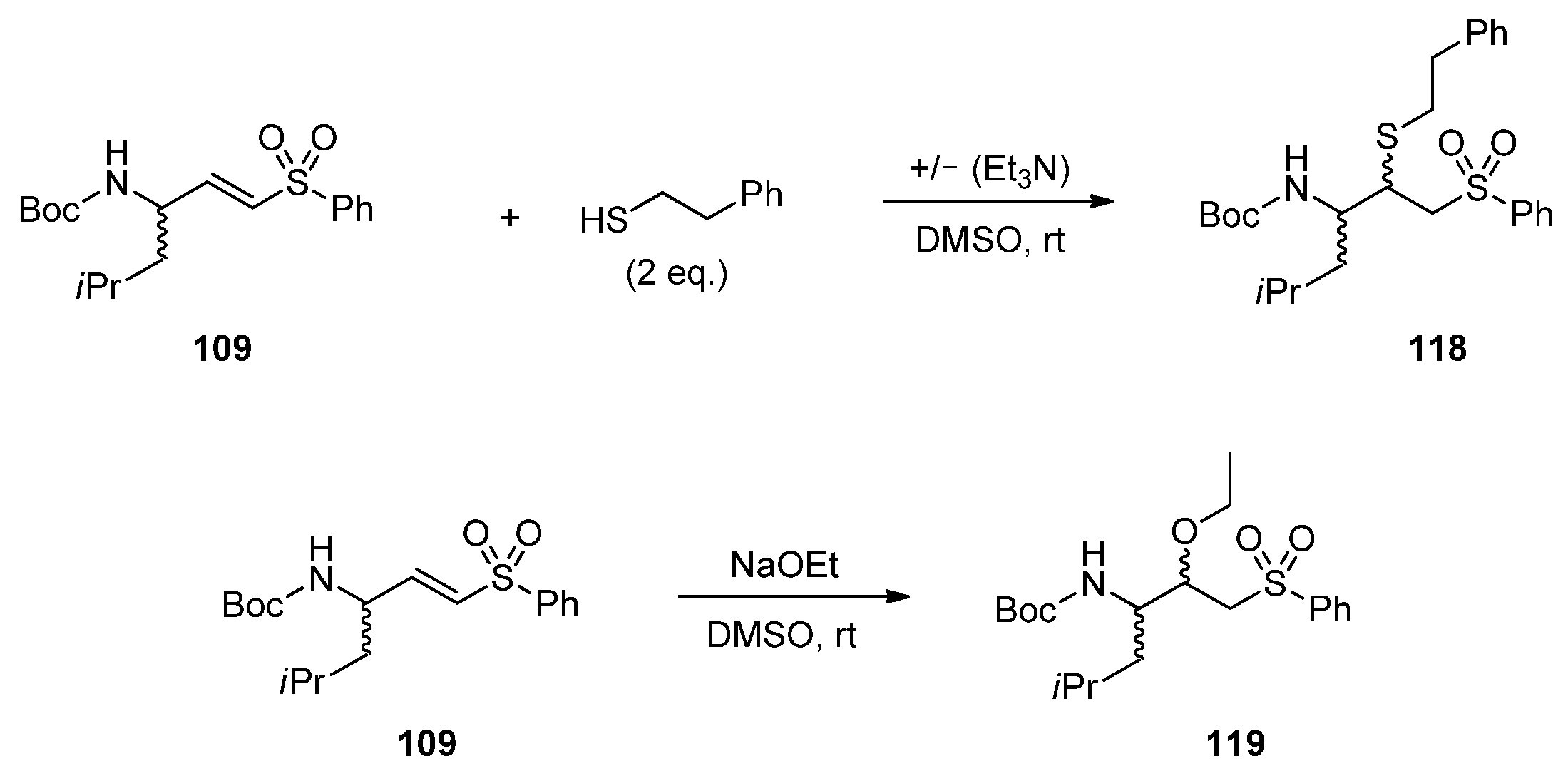
2.1.7. Synthesis of Reactivity Probes
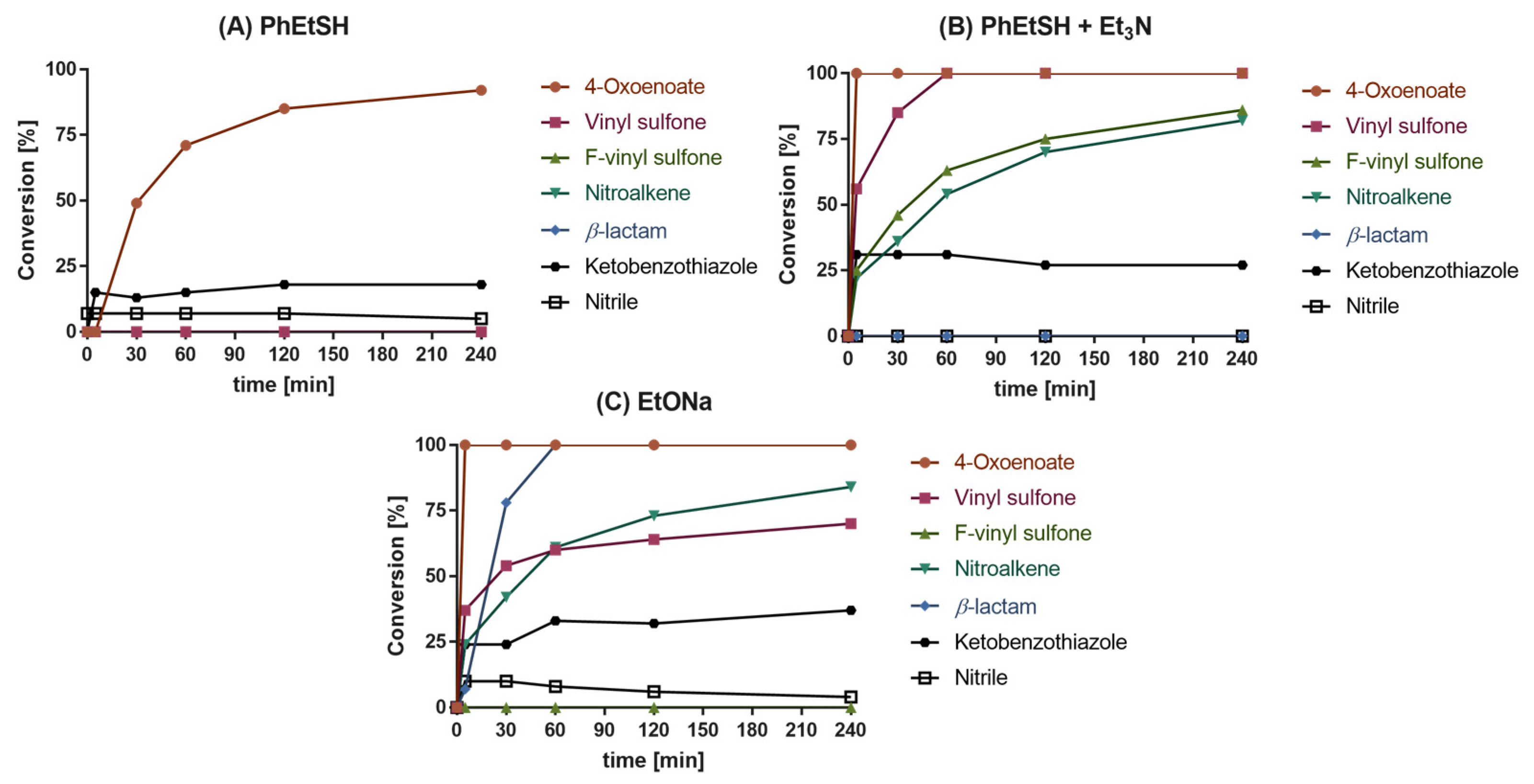
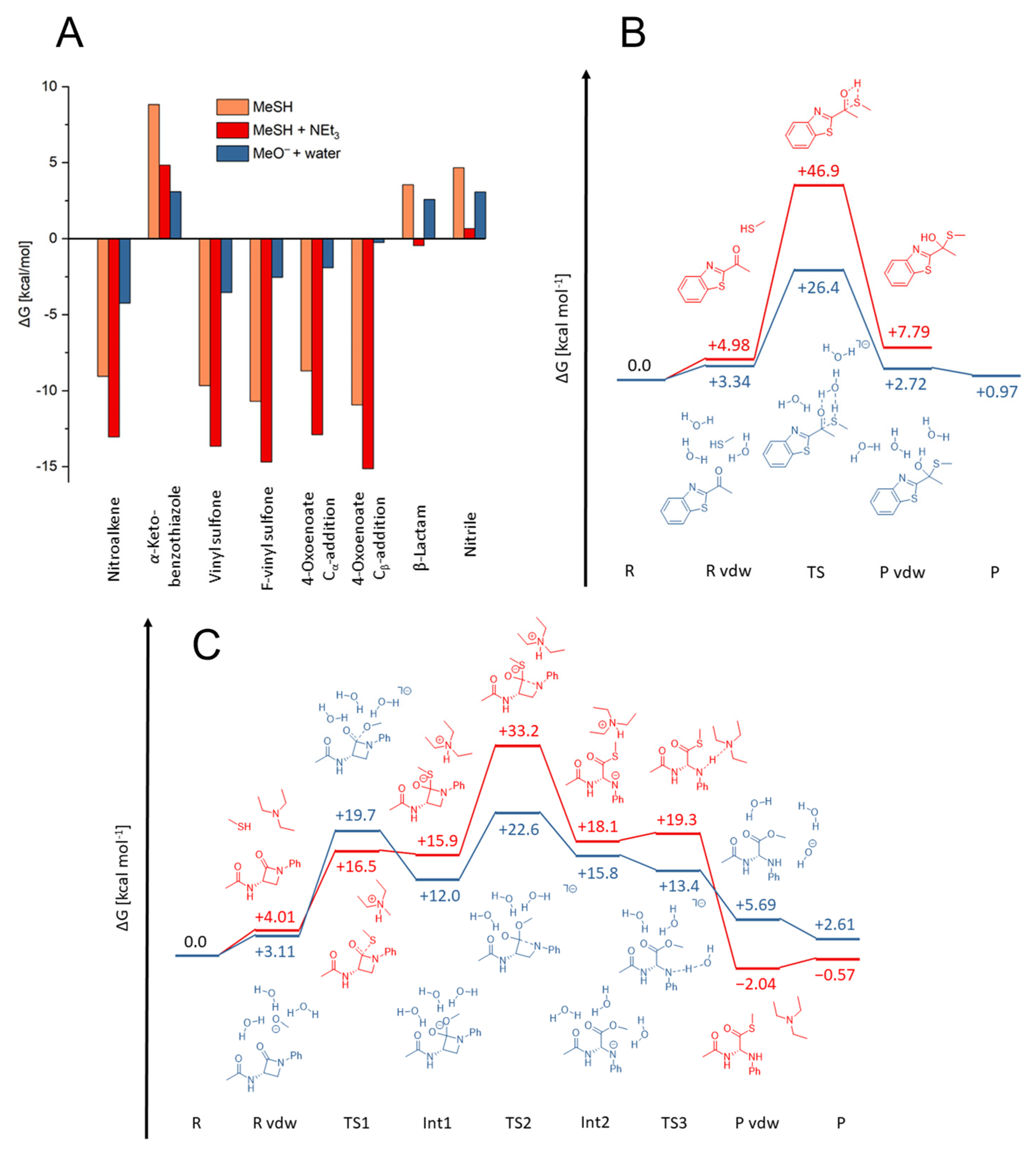
2.2. Reactivity Tests
2.3. Quantum Mechanics Simulations
2.4. In Vitro Evaluation of the Synthesized Compounds
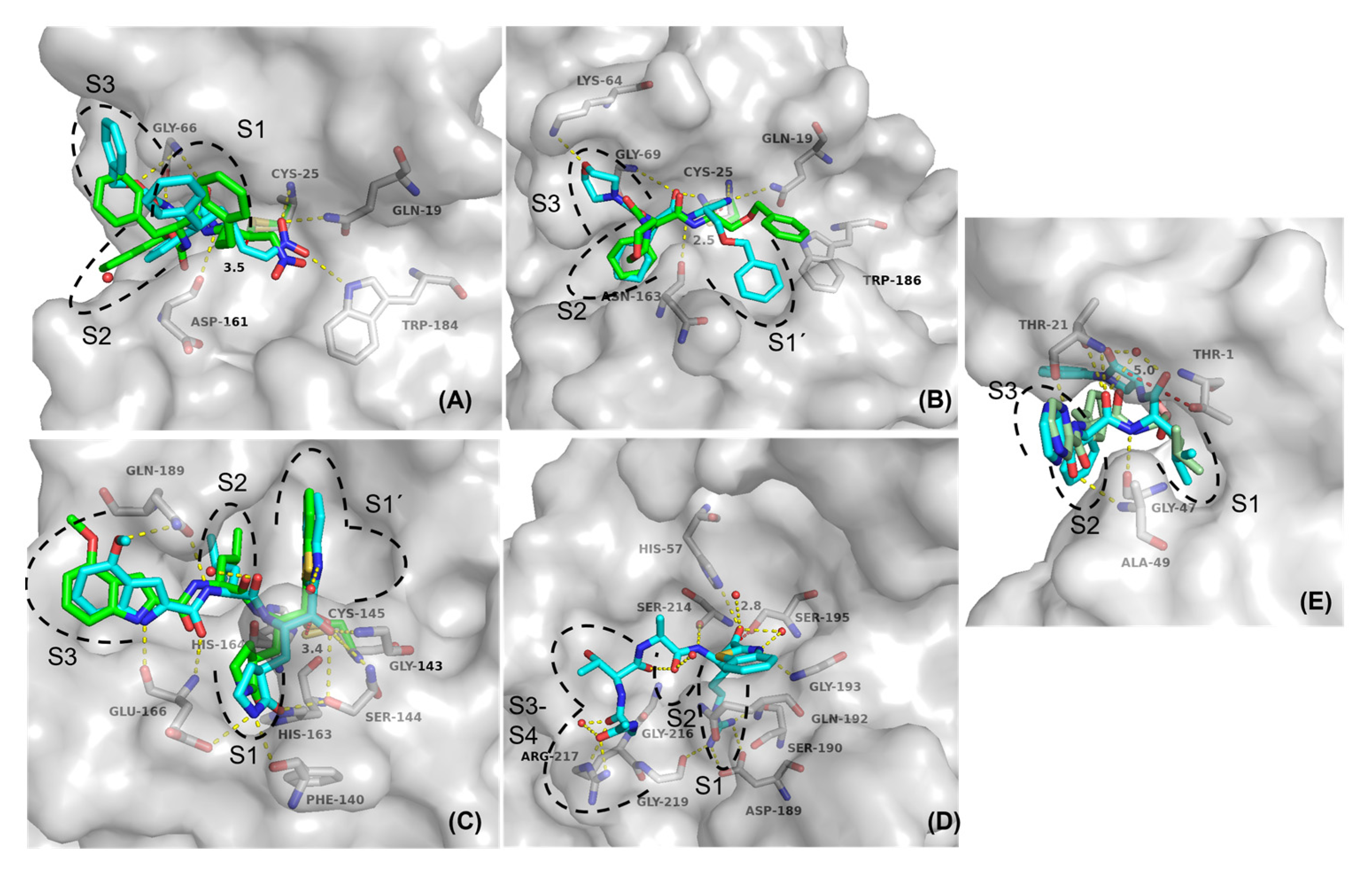
2.5. Molecular Docking
2.6. Comparison of the Reactivity Assay Results with the In Vitro Study
3. Discussion
4. Material and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Overall, C.M. Protease degradomics: A new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002, 3, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Grozdanić, M.; Vidmar, R.; Vizovišek, M.; Fonović, M. Degradomics in Biomarker Discovery. Proteom. Clin. Appl. 2019, 13, 1800138. [Google Scholar] [CrossRef] [PubMed]
- Ruggiano, A.; Ramadan, K. DNA–protein crosslink proteases in genome stability. Commun. Biol. 2021, 4, 11. [Google Scholar] [CrossRef]
- Lee, C.W.; Stankowski, J.N.; Chew, J.; Cook, C.N.; Lam, Y.W.; Almeida, S.; Carlomagno, Y.; Lau, K.F.; Prudencio, M.; Gao, F.B.; et al. The lysosomal protein cathepsin L is a progranulin protease. Mol. Neurodegener. 2017, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eatemadi, A.; Aiyelabegan, H.T.; Negahdari, B.; Mazlomi, M.A.; Daraee, H.; Daraee, N.; Eatemadi, R.; Sadroddiny, E. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed. Pharmacother. 2017, 86, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Guo, J.; Zhang, X.; Sukhova, G.K.; Libby, P.; Shi, G.P. Cysteine protease cathepsins in cardiovascular disease: From basic research to clinical trials. Nat. Rev. Cardiol. 2018, 15, 351–370. [Google Scholar] [CrossRef]
- Previti, S.; Ettari, R.; Calcaterra, E.; Di Chio, C.; Ravichandran, R.; Zimmer, C.; Hammerschmidt, S.; Wagner, A.; Bogacz, M.; Cosconati, S.; et al. Development of Urea-Bond-Containing Michael Acceptors as Antitrypanosomal Agents Targeting Rhodesain. ACS Med. Chem. Lett. 2022, 13, 1083–1090. [Google Scholar] [CrossRef]
- Rocha, D.A.; Silva, E.B.; Fortes, I.S.; Lopes, M.S.; Ferreira, R.S.; Andrade, S.F. Synthesis and structure-activity relationship studies of cruzain and rhodesain inhibitors. Eur. J. Med. Chem. 2018, 157, 1426–1459. [Google Scholar] [CrossRef]
- Kincaid, J.R.A.; Caravez, J.C.; Iyer, K.S.; Kavthe, R.D.; Fleck, N.; Aue, D.H.; Lipshutz, B.H. A sustainable synthesis of the SARS-CoV-2 Mpro inhibitor nirmatrelvir, the active ingredient in Paxlovid. Commun. Chem. 2022, 5, 156. [Google Scholar] [CrossRef]
- Müller, P.; Maus, H.; Hammerschmidt, S.J.; Knaff, P.M.; Mailänder, V.; Schirmeister, T.; Kersten, C. Interfering with Host Proteases in SARS-CoV-2 Entry as a Promising Therapeutic Strategy. Curr. Med. Chem. 2022, 29, 635–665. [Google Scholar] [CrossRef]
- Knaff, P.M.; Müller, P.; Kersten, C.; Wettstein, L.; Münch, J.; Landfester, K.; Mailänder, V. Structure-Based Design of High-Affinity and Selective Peptidomimetic Hepsin Inhibitors. Biomacromolecules 2022, 23, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Tsantrizos, Y.S.; Bolger, G.; Bonneau, P.; Cameron, D.R.; Goudreau, N.; Kukolj, G.; LaPlante, S.R.; Llinàs-Brunet, M.; Nar, H.; Lamarre, D. Macrocyclic inhibitors of the NS3 protease as potential therapeutic agents of hepatitis C virus infection. Angew. Chemie Int. Ed. 2003, 42, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Maus, H.; Barthels, F.; Hammerschmidt, S.J.; Kopp, K.; Millies, B.; Gellert, A.; Ruggieri, A.; Schirmeister, T. SAR of novel benzothiazoles targeting an allosteric pocket of DENV and ZIKV NS2B/NS3 proteases. Bioorg. Med. Chem. 2021, 47, 116392. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorg. Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Zhang, F.; Diao, H.; Wu, R. Covalent Inhibition Mechanism of Antidiabetic Drugs—Vildagliptin vs Saxagliptin. ACS Catal. 2019, 9, 2292–2302. [Google Scholar] [CrossRef]
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D 2019, 19, 73–92. [Google Scholar] [CrossRef] [Green Version]
- Johe, P.; Jung, S.; Endres, E.; Kersten, C.; Zimmer, C.; Ye, W.; Sönnichsen, C.; Hellmich, U.A.; Sotriffer, C.; Schirmeister, T.; et al. Warhead Reactivity Limits the Speed of Inhibition of the Cysteine Protease Rhodesain. ACS Chem. Biol. 2021, 16, 661–670. [Google Scholar] [CrossRef]
- Santos, M.; Moreira, R. Mini-Reviews. Med. Chem. 2007, 7, 1040–1050. [Google Scholar]
- Adams, J.; Kauffman, M. Development of the Proteasome Inhibitor VelcadeTM (Bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef]
- Dražić, T.; Kopf, S.; Corridan, J.; Leuthold, M.M.; Bertoša, B.; Klein, C.D. Peptide-β-lactam Inhibitors of Dengue and West Nile Virus NS2B-NS3 Protease Display Two Distinct Binding Modes. J. Med. Chem. 2020, 63, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, T.L.; Breddam, K.; Remington, J.S. Peptide Aldehyde Complexes with Wheat Serine Carboxypeptidase II: Implications for the Catalytic Mechanism and Substrate Specificity. J. Mol. Biol. 1996, 255, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lin, C.; Xu, Q.; Zhou, X.; Zeng, P.; McCormick, P.J.; Jiang, H.; Li, J.; Zhang, J. Structural Basis for the Inhibition of Coronaviral Main Proteases by a Benzothiazole-Based Inhibitor. Viruses 2022, 14, 2075. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Tsutsumi, S.; Hatsushiba, E.; Ohuchi, S.; Okonogi, T. Peptidyl α-keto thiazole as potent thrombin inhibitors. Bioorg. Med. Chem. Lett. 1997, 7, 533–538. [Google Scholar] [CrossRef]
- Vincenza Carriero, M.; Patrizia Stoppelli, M. The Urokinase-type Plasminogen Activator and the Generation of Inhibitors of Urokinase Activity and Signaling. Curr. Pharm. Des. 2011, 17, 1944–1961. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.A.; Shaker, B.T.; Bajou, K. The plasminogen–activator plasmin system in physiological and pathophysiological angiogenesis. Int. J. Mol. Sci. 2021, 23, 337. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Kumar, A.A.; Buckley, B.J.; Ranson, M. The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets. Biomolecules 2022, 12, 152. [Google Scholar] [CrossRef]
- Li, C.Y.; de Veer, S.J.; Law, R.H.P.; Whisstock, J.C.; Craik, D.J.; Swedberg, J.E. Characterising the Subsite Specificity of Urokinase-Type Plasminogen Activator and Tissue-Type Plasminogen Activator using a Sequence-Defined Peptide Aldehyde Library. ChemBioChem 2019, 20, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome Inhibitors: An Expanding Army Attacking a Unique Target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, I.D.; Wu, P.; Marion-Tsukamaki, R.; Mackey, Z.B.; Brinen, L.S. Crystal Structures of TbCatB and Rhodesain, Potential Chemotherapeutic Targets and Major Cysteine Proteases of Trypanosoma brucei. PLoS Negl. Trop. Dis. 2010, 4, e701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauly, T.A.; Sulea, T.; Ammirati, M.; Sivaraman, J.; Danley, D.E.; Griffor, M.C.; Kamath, A.V.; Wang, I.K.; Laird, E.R.; Seddon, A.P.; et al. Specificity determinants of human cathepsin S revealed by crystal structures of complexes. Biochemistry 2003, 42, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.D.A.; Williams, R.; Scott, C.J.; Burden, R.E. Cathepsin S: Therapeutic, diagnostic, and prognostic potential. Biol. Chem. 2015, 396, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-C.; Uang, B.-J.; Lyu, P.-C.; Chang, J.-Y.; Liu, K.-J.; Kuo, C.-C.; Hsieh, H.-P.; Wang, H.-C.; Cheng, C.-S.; Chang, Y.-H.; et al. Design and Synthesis of α-Ketoamides as Cathepsin S Inhibitors with Potential Applications against Tumor Invasion and Angiogenesis. J. Med. Chem. 2010, 53, 4545–4549. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Fuchs, N.; Johe, P.; Wagner, A.; Diehl, E.; Yuliani, T.; Zimmer, C.; Barthels, F.; Zimmermann, R.A.; Klein, P.; et al. Fluorovinylsulfones and -Sulfonates as Potent Covalent Reversible Inhibitors of the Trypanosomal Cysteine Protease Rhodesain: Structure-Activity Relationship, Inhibition Mechanism, Metabolism, and in Vivo Studies. J. Med. Chem. 2021, 64, 12322–12358. [Google Scholar] [CrossRef]
- Previti, S.; Ettari, R.; Cosconati, S.; Schirmeister, G.; Chouchene, K.; Wagner, A.; Hellmich, U.A.; Ulrich, K.; Krauth-Siegel, R.L.; Wich, P.R.; et al. Development of Novel Peptide-Based Michael Acceptors Targeting Rhodesain and Falcipain-2 for the Treatment of Neglected Tropical Diseases (NTDs). J. Med. Chem. 2017, 60, 6911–6923. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Zeslawska, E.; Jacob, U.; Schweinitz, A.; Coombs, G.; Bode, W.; Madison, E. Crystals of urokinase type plasminogen activator complexes reveal the binding mode of peptidomimetic inhibitors. J. Mol. Biol. 2003, 328, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, Y.D.; Thomson, D.S.; Frye, L.L.; Cywin, C.L.; Morwick, T.; Emmanuel, M.J.; Zindell, R.; McNeil, D.; Bekkali, Y.; Marc Girardot, M.; et al. Design and synthesis of dipeptide nitriles as reversible and potent Cathepsin S inhibitors. J. Med. Chem. 2002, 45, 5471–5482. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.-I.; Higashi-Kuwata, N.; Hayashi, H.; Allu, S.R.; Raghavaiah, J.; Bulut, H.; Das, D.; Anson, B.J.; Lendy, E.K.; Takamatsu, Y.; et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2021, 12, 668. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, W.; Vederas, J.C. Improved Synthesis of a Cyclic Glutamine Analogue Used in Antiviral Agents Targeting 3C and 3CL Proteases Including SARS-CoV-2 M pro. J. Org. Chem. 2021, 86, 13104–13110. [Google Scholar] [CrossRef]
- Tian, Q.; Nayyar, N.K.; Babu, S.; Chen, L.; Tao, J.; Lee, S.; Tibbetts, A.; Moran, T.; Liou, J.; Guo, M.; et al. An efficient synthesis of a key intermediate for the preparation of the rhinovirus protease inhibitor AG7088 via asymmetric dianionic cyanomethylation of N-Boc-l-(+)-glutamic acid dimethyl ester. Tetrahedron Lett. 2001, 42, 6807–6809. [Google Scholar] [CrossRef]
- Royo, S.; Rodríguez, S.; Schirmeister, T.; Kesselring, J.; Kaiser, M.; González, F.V. Dipeptidyl Enoates As Potent Rhodesain Inhibitors That Display a Dual Mode of Action. ChemMedChem 2015, 10, 1484–1487. [Google Scholar] [CrossRef]
- Latorre, A.; Schirmeister, T.; Kesselring, J.; Jung, S.; Johé, P.; Hellmich, U.A.; Heilos, A.; Engels, B.; Krauth-Siegel, R.L.; Dirdjaja, N.; et al. Dipeptidyl Nitroalkenes as Potent Reversible Inhibitors of Cysteine Proteases Rhodesain and Cruzain. ACS Med. Chem. Lett. 2016, 7, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.K.; Finke, P.E.; Brause, K.A.; Chandler, G.O.; Ashe, B.M.; Weston, H.; Maycock, A.L.; Mumford, R.A.; Doherty, J.B. Monocyclic β-lactam inhibitors of human leukocyte elastase. Stereospecific synthesis and activity of 3,4-disubstituted-2-azetidinones. Bioorg. Med. Chem. Lett. 1993, 3, 2295–2298. [Google Scholar] [CrossRef]
- Han, W.T.; Trehan, A.K.; Kim Wright, J.J.; Federici, M.E.; Seiler, S.M.; Meanwell, N.A. Azetidin-2-one derivatives as inhibitors of thrombin. Bioorg. Med. Chem. 1995, 3, 1123–1143. [Google Scholar] [CrossRef] [PubMed]
- Steert, K.; Berg, M.; Mottram, J.C.; Westrop, G.D.; Coombs, G.H.; Cos, P.; Maes, L.; Joossens, J.; Van der Veken, P.; Haemers, A.; et al. α-Ketoheterocycles as Inhibitors of Leishmania mexicana Cysteine Protease CPB. ChemMedChem 2010, 5, 1734–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzo, M.J.; Almond, H.R.; Hecker, L.R.; Schott, M.R.; Yabut, S.C.; Zhang, H.-C.; Andrade-Gordon, P.; Corcoran, T.W.; Giardino, E.C.; Kauffman, J.A.; et al. In-Depth Study of Tripeptide-Based α-Ketoheterocycles as Inhibitors of Thrombin. Effective Utilization of the S1 ‘Subsite and Its Implications to Structure-Based Drug Design. J. Med. Chem. 2005, 48, 1984–2008. [Google Scholar] [CrossRef] [PubMed]
- DiNinno, F.; Ernest, V.L. Facile Synthesis of β-Thioxo Esters from β-Enamino Esters. J. Org. Chem. 1979, 44, 3271–3273. [Google Scholar] [CrossRef]
- Delprino, L.; Giacomotti, M.; Dosio, F.; Brusa, P.; Ceruti, M.; Grosa, G.; Cattel, L. Toxin-Targeted Design for Anticancer Therapy. I: Synthesis and Biological Evaluation of New Thioimidate Heterobifunctional Reagents. J. Pharm. Sci. 1993, 82, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Ibba, R.; Rossi, S.; Butini, S.; Calderone, V.; Gemma, S.; Campiani, G. Covalent Reversible Inhibitors of Cysteine Proteases Containing the Nitrile Warhead: Recent Advancement in the Field of Viral and Parasitic Diseases. Molecules 2022, 27, 2561. [Google Scholar] [CrossRef]
- Barthels, F.; Meyr, J.; Hammerschmidt, S.J.; Marciniak, T.; Räder, H.-J.; Ziebuhr, W.; Engels, B.; Schirmeister, T. 2-Sulfonylpyrimidines as Privileged Warheads for the Development of S. aureus Sortase A Inhibitors. Front. Mol. Biosci. 2022, 8, 804970. [Google Scholar] [CrossRef]
- Paasche, A.; Schiller, M.; Schirmeister, T.; Engels, B. Mechanistic Study of the Reaction of Thiol-Containing Enzymes with α,β-Unsaturated Carbonyl Substrates by Computation and Chemoassays. ChemMedChem 2010, 5, 869–880. [Google Scholar] [CrossRef]
- Silva, D.G.; Ribeiro, J.F.R.; De Vita, D.; Cianni, L.; Franco, C.H.; Freitas-Junior, L.H.; Moraes, C.B.; Rocha, J.R.; Burtoloso, A.C.B.; Kenny, P.W.; et al. A comparative study of warheads for design of cysteine protease inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5031–5035. [Google Scholar] [CrossRef]
- Kim, K.B.; Crews, C.M. From epoxomicin to carfilzomib: Chemistry, biology, and medical outcomes. Nat. Prod. Rep. 2013, 30, 600. [Google Scholar] [CrossRef] [Green Version]
- Cianni, L.; Feldmann, C.W.; Gilberg, E.; Gütschow, M.; Juliano, L.; Leitão, A.; Bajorath, J.; Montanari, C.A. Can Cysteine Protease Cross-Class Inhibitors Achieve Selectivity? J. Med. Chem. 2019, 62, 10497–10525. [Google Scholar] [CrossRef] [PubMed]
- Konno, S.; Kobayashi, K.; Senda, M.; Funai, Y.; Seki, Y.; Tamai, I.; Schäkel, L.; Sakata, K.; Pillaiyar, T.; Taguchi, A.; et al. 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem. 2022, 65, 2926–2939. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2020; Available online: https://www.chemcomp.com/. (accessed on 23 March 2022).
- LeadIT/FlexX, Version 2.3.2; GmbH, BioSolveIT: Sankt Augustin, Germany, 2017.
- Serrano-Aparicio, N.; Moliner, V.; Świderek, K. Nature of Irreversible Inhibition of Human 20S Proteasome by Salinosporamide A. The Critical Role of Lys–Asp Dyad Revealed from Electrostatic Effects Analysis. ACS Catal. 2021, 11, 3575–3589. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Chenna, B.C.; Li, L.; Mellott, D.M.; Zhai, X.; Siqueira-Neto, J.L.; Calvet Alvarez, C.; Bernatchez, J.A.; Desormeaux, E.; Alvarez Hernandez, E.; Gomez, J.; et al. Peptidomimetic Vinyl Heterocyclic Inhibitors of Cruzain Effect Antitrypanosomal Activity. J. Med. Chem. 2020, 63, 3298–3316. [Google Scholar] [CrossRef]
- Vankadara, S.; Dawson, M.D.; Fong, J.Y.; Oh, Q.Y.; Ang, Q.A.; Liu, B.; Chang, H.Y.; Koh, J.; Koh, X.; Tan, Q.W.; et al. A Warhead Substitution Study on the Coronavirus Main Protease Inhibitor Nirmatrelvir. ACS Med. Chem. Lett. 2022, 13, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Ludewig, S.; Kossner, M.; Schiller, M.; Baumann, K.; Schirmeister, T. Enzyme Kinetics and Hit Validation in Fluorimetric Protease Assays. Curr. Top. Med.Chem. 2010, 10, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Barthels, F.; Marincola, G.; Marciniak, T.; Konhäuser, M.; Hammerschmidt, S.; Bierlmeier, J.; Distler, U.; Wich, P.R.; Tenzer, S.; Schwarzer, D.; et al. Irreversible and Selective Inhibitors of Staphylococcus aureus Sortase A. ChemMedChem 2020, 15, 839–850. [Google Scholar] [CrossRef]
- Amendola, G.; Ettari, R.; Previti, S.; Di Chio, C.; Messere, A.; Di Maro, S.S.; Hammerschmidt, J.; Zimmer, C.; Zimmermann, R.A.; Schirmeister, T.; et al. Lead Discovery of SARS-CoV-2 Main Protease Inhibitors through Covalent Docking-Based Virtual Screening. J. Chem. Inf. Model. 2021, 61, 2062–2073. [Google Scholar] [CrossRef]
- Schirmeister, T.; Kesselring, J.; Jung, S.; Schneider, T.H.; Weickert, A.; Becker, J.; Lee, W.; Bamberger, D.; Wich, P.R.; Distler, U.; et al. Engels, Quantum Chemical-Based Protocol for the Rational Design of Covalent Inhibitors. J. Am. Chem. Soc. 2016, 138, 8332–8335. [Google Scholar] [CrossRef]
- Caffrey, C.R.; Hansell, E.; Lucas, K.D.; Brinen, L.S.; Hernandez, A.A.; Cheng, J.; Roush, W.R.; Stierhof, Y.-D.; Bogyo, M.; Steverding, D.; et al. Active site mapping, biochemical properties and subcellular localization of rhodesain, the major cysteine protease of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 2001, 118, 61–73. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 2.5.2; Schrödinger, LLC: New York, NY, USA, 2021.
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Peterson, G.A.; Nakatsuji, H.; et al. Gaussian 16 (Revision A.03); Gaussian Inc.: Wallingfort, CT, USA, 2016. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Pliego, J.R., Jr.; Riveros, J.M. Gibbs energy of solvation of organic ions in aqueous and dimethyl sulfoxide solutions. Phys. Chem. Chem. Phys. 2002, 4, 1622–1627. [Google Scholar] [CrossRef]
- Ben-Naim, A. Standard Thermodynamics of Transfer. Uses and Misuses. J. Phys. Chem. 1978, 82, 792–803. [Google Scholar] [CrossRef]
- Spina, R.; Colacino, E.; Martinez, J.; Lamaty, F. Poly(ethylene glycol) as a Reaction Matrix in Platinum- or Gold-Catalyzed Cycloisomerization: A Mechanistic Investigation. Chem.-A Eur. J. 2013, 19, 3817–3821. [Google Scholar] [CrossRef]
- Ho, A.; Cyrus, K.; Kim, K.-B. Towards Immunoproteasome-Specific Inhibitors: An Improved Synthesis of Dihydroeponemycin. Eur. J. Org. Chem. 2005, 2005, 4829–4834. [Google Scholar] [CrossRef]
- St-Georges, C.; Désilets, A.; Béliveau, F.; Ghinet, M.; Dion, S.P.; Colombo, É.; Boudreault, P.-L.; Najmanovich, R.J.; Leduc, R.; Marsault, É. Modulating the selectivity of matriptase-2 inhibitors with unnatural amino acids. Eur. J. Med. Chem. 2017, 129, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.J.; Yabut, S.C.; Almond, H.R.; Andrade-Gordon, P.; Corcoran, T.W.; de Garavilla, L.; Kauffman, J.A.; Abraham, W.M.; Recacha, R.; Chattopadhyay, D.; et al. Potent, Small-Molecule Inhibitors of Human Mast Cell Tryptase. Antiasthmatic Action of a Dipeptide-Based Transition-State Analogue Containing a Benzothiazole Ketone. J. Med. Chem. 2003, 46, 3865–3876. [Google Scholar] [CrossRef]
- Engel-Andreasen, J.; Wellhöfer, I.; Wich, K.; Olsen, C.A. Backbone-Fluorinated 1,2,3-Triazole-Containing Dipeptide Surrogates. J. Org. Chem. 2017, 82, 11613–11619. [Google Scholar] [CrossRef] [PubMed]
- Dutton, F.E.; Lee, B.H.; Johnson, S.S.; Coscarelli, E.M.; Lee, P.H. Restricted Conformation Analogues of an Anthelmintic Cyclodepsipeptide. J. Med. Chem. 2003, 46, 2057–2073. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, P.; Meta, M.; Meidner, J.L.; Schwickert, M.; Meyr, J.; Schwickert, K.; Kersten, C.; Zimmer, C.; Hammerschmidt, S.J.; Frey, A.; et al. Investigation of the Compatibility between Warheads and Peptidomimetic Sequences of Protease Inhibitors—A Comprehensive Reactivity and Selectivity Study. Int. J. Mol. Sci. 2023, 24, 7226. https://doi.org/10.3390/ijms24087226
Müller P, Meta M, Meidner JL, Schwickert M, Meyr J, Schwickert K, Kersten C, Zimmer C, Hammerschmidt SJ, Frey A, et al. Investigation of the Compatibility between Warheads and Peptidomimetic Sequences of Protease Inhibitors—A Comprehensive Reactivity and Selectivity Study. International Journal of Molecular Sciences. 2023; 24(8):7226. https://doi.org/10.3390/ijms24087226
Chicago/Turabian StyleMüller, Patrick, Mergim Meta, Jan Laurenz Meidner, Marvin Schwickert, Jessica Meyr, Kevin Schwickert, Christian Kersten, Collin Zimmer, Stefan Josef Hammerschmidt, Ariane Frey, and et al. 2023. "Investigation of the Compatibility between Warheads and Peptidomimetic Sequences of Protease Inhibitors—A Comprehensive Reactivity and Selectivity Study" International Journal of Molecular Sciences 24, no. 8: 7226. https://doi.org/10.3390/ijms24087226
APA StyleMüller, P., Meta, M., Meidner, J. L., Schwickert, M., Meyr, J., Schwickert, K., Kersten, C., Zimmer, C., Hammerschmidt, S. J., Frey, A., Lahu, A., de la Hoz-Rodríguez, S., Agost-Beltrán, L., Rodríguez, S., Diemer, K., Neumann, W., Gonzàlez, F. V., Engels, B., & Schirmeister, T. (2023). Investigation of the Compatibility between Warheads and Peptidomimetic Sequences of Protease Inhibitors—A Comprehensive Reactivity and Selectivity Study. International Journal of Molecular Sciences, 24(8), 7226. https://doi.org/10.3390/ijms24087226