
The Role of Platelets in Infective Endocarditis


 ,
, 
Abstract
:1. Introduction
2. Platelet Involvement in the Pathophysiology of Infectious Endocarditis

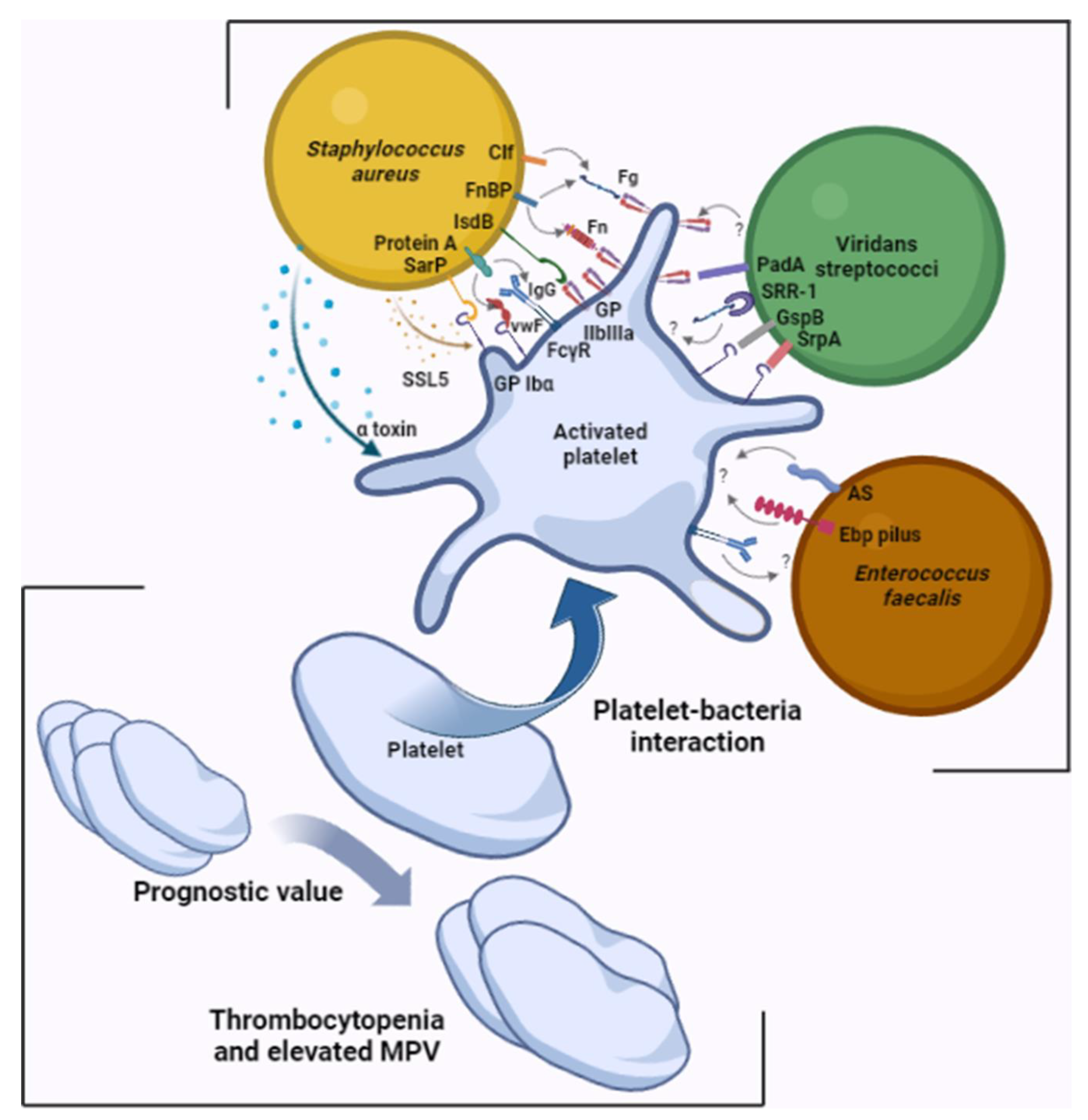{kind=link}
{kind=link}
| Bacterial Surface Molecule | Plasma Protein | Platelet | Reference |
|---|---|---|---|
| Staphylococcus aureus | |||
| IsdB | - | GPIIbIIIa | [14] |
| Sar P | - | GPIbα | [15] |
| Protein A | IgG | FcγRII | [16] |
| VWF | GPIbα | [17] | |
| Clf A and Clf B | Fg and Fn | GPIIbIIIa | [18,19,20,21] |
| FnBP | |||
| Viridans streptococci | |||
| SrpA | - | GPIbα | [26] |
| GspB | - | GPIbα | [27] |
| PadA | - | GPIIbIIIa | [28] |
| SRR-1 | Fg | ? | [29] |
| Enterococcus faecalis | |||
| AS | - | ? | [30] |
| Ebp | - | ? | [31] |
3. Platelets Are Key Actor in the Formation of Vegetation
4. Involvement of Platelets in Antibacterial Immunity
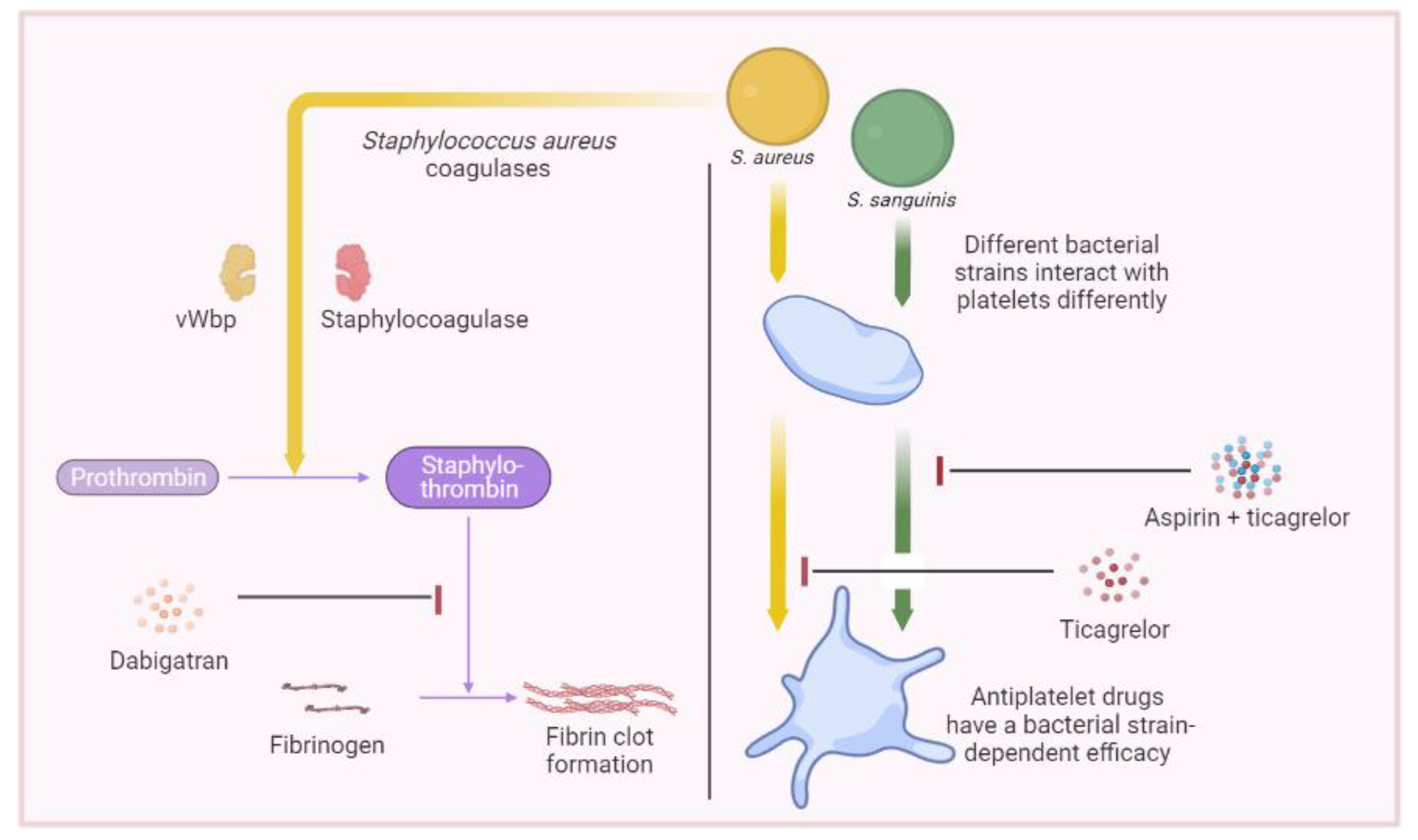
5. Therapeutics Involving Platelets
6. Major Research Techniques Used for Platelet–Bacteria Interaction Studies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cahill, T.J.; Baddour, L.M.; Habib, G.; Hoen, B.; Salaun, E.; Pettersson, G.B.; Schäfers, H.J.; Prendergast, B.D. Challenges in infective endocarditis. J. Am. Coll. Cardiol. 2017, 69, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Habib, G.; Lancellotti, P.; Erba, P.A.; Sadeghpour, A.; Meshaal, M.; Sambola, A.; Furnaz, S.; Citro, R.; Ternacle, J.; Donal, E.; et al. The ESC-EORP EURO-ENDO (European Infective Endocarditis) registry. Eur. Heart J. Qual. Care Clin. Outcomes 2019, 5, 202–207, Erratum in Eur. Heart J. Qual. Care Clin. Outcomes 2020, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Cabiltes, I.; Coghill, S.; Bowe, S.J.; Athan, E. Enterococcal bacteraemia ‘silent but deadly’: A population-based cohort study. Intern. Med. J. 2020, 50, 434–440. [Google Scholar] [CrossRef]
- Beganovic, M.; Luther, M.K.; Rice, L.B.; Arias, C.A.; Rybak, M.J.; LaPlante, K.L. A Review of Combination Antimicrobial Therapy for Enterococcus faecalis Bloodstream Infections and Infective Endocarditis. Clin. Infect. Dis. 2018, 67, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Pazdernik, M.; Iung, B.; Mutlu, B.; Alla, F.; Riezebos, R.; Kong, W.; Nunes, M.C.P.; Pierard, L.; Srdanovic, I.; Yamada, H.; et al. Surgery and outcome of infective endocarditis in octogenarians: Prospective data from the ESC EORP EURO-ENDO registry. Infection 2022, 50, 1191–1202. [Google Scholar] [CrossRef]
- Bouza, E.; Muñoz, P.; Burillo, A. Gram-negative endocarditis: Disease presentation, diagnosis and treatment. Curr. Opin. Infect. Dis. 2021, 34, 672–680. [Google Scholar] [CrossRef]
- Charlesworth, M.; Williams, B.G.; Ray, S. Infective endocarditis. BJA Educ. 2023, 23, 144–152. [Google Scholar] [CrossRef]
- Alonso, B.; Pérez-Granda, M.J.; Latorre, M.C.; Sánchez-Carrillo, C.; Bouza, E.; Muñoz, P.; Guembe, M. Production of biofilm by Staphylococcus aureus: Association with infective endocarditis? Enferm. Infecc. Microbiol. Clin. (Engl. Ed.) 2022, 40, 418–422. [Google Scholar] [CrossRef]
- Kouijzer, J.J.P.; Noordermeer, D.J.; van Leeuwen, W.J.; Verkaik, N.J.; Lattwein, K.R. Native valve, prosthetic valve, and cardiac device-related infective endocarditis: A review and update on current innovative diagnostic and therapeutic strategies. Front. Cell Dev. Biol. 2022, 10, 995508. [Google Scholar] [CrossRef]
- Deppermann, C.; Kubes, P. Start a fire, kill the bug: The role of platelets in inflammation and infection. Innate Immun. 2018, 24, 335–348. [Google Scholar] [CrossRef]
- Sharda, A.; Flaumenhaft, R. The life cycle of platelet granules. F1000Research 2018, 7, 236. [Google Scholar] [CrossRef]
- Patel, S.R.; Hartwig, J.H.; Italiano, J.E., Jr. The biogenesis of platelets from megakaryocyte proplatelets. J. Clin. Investig. 2005, 115, 3348–3354. [Google Scholar] [CrossRef] [PubMed]
- Hannachi, N.; Habib, G.; Camoin-Jau, L. Aspirin Effect on Staphylococcus aureus-Platelet Interactions During Infectious Endocarditis. Front. Med. 2019, 6, 217. [Google Scholar] [CrossRef] [PubMed]
- Miajlovic, H.; Zapotoczna, M.; Geoghegan, J.A.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology 2010, 156, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Siboo, I.R.; Chambers, H.F.; Sullam, P.M. Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect. Immun. 2005, 73, 2273–2280. [Google Scholar] [CrossRef]
- Hawiger, J.; Steckley, S.; Hammond, D.; Cheng, C.; Timmons, S.; Glick, A.D.; Prez, R.M.D. Staphylococci-induced human platelet injury mediated by protein A and immunoglobulin G Fc fragment receptor. J. Clin. Investig. 1979, 64, 931–937. [Google Scholar] [CrossRef]
- O’Seaghdha, M.; van Schooten, C.J.; Kerrigan, S.W.; Emsley, J.; Silverman, G.J.; Cox, D.; Lenting, P.J.; Foster, T.J. Staphylococcus aureus protein A binding to von Willebrand factor A1 domain is mediated by conserved IgG binding regions. FEBS J. 2006, 273, 4831–4841. [Google Scholar] [CrossRef]
- Ní Eidhin, D.; Perkins, S.; Francois, P.; Vaudaux, P.; Höök, M.; Foster, T.J. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol. Microbiol. 1998, 30, 245–257. [Google Scholar] [CrossRef]
- Entenza, J.M.; Foster, T.J.; Ni Eidhin, D.; Vaudaux, P.; Francioli, P.; Moreillon, P. Contribution of clumping factor B to pathogenesis of experimental endocarditis due to Staphylococcus aureus. Infect. Immun. 2000, 68, 5443–5446. [Google Scholar] [CrossRef]
- Liu, C.-Z.; Huang, T.-F.; Tsai, P.-J.; Tsai, P.-J.; Chang, L.-Y.; Chang, M.-C. A segment of Staphylococcus aureus clumping factor A with fibrinogen-binding activity (ClfA221-550) inhibits platelet-plug formation in mice. Thromb. Res. 2007, 121, 183–191. [Google Scholar] [CrossRef]
- Foster, T.J. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Surewaard, B.G.J.; Thanabalasuriar, A.; Zeng, Z.; Tkaczyk, C.; Cohen, T.S.; Bardoel, B.W.; Jorch, S.K.; Deppermann, C.; Wardenburg, J.B.; Davis, R.P.; et al. α-Toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis. Cell Host. Microbe 2018, 24, 271–284.e3. [Google Scholar] [CrossRef] [PubMed]
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Peetermans, M.; Vanassche, T.; Liesenborghs, L.; Lijnen, R.H.; Verhamme, P. Bacterial pathogens activate plasminogen to breach tissue barriers and escape from innate immunity. Crit. Rev. Microbiol. 2016, 42, 866–882. [Google Scholar] [CrossRef] [PubMed]
- Ford, I.; Douglas, C.W.; Preston, F.E.; Lawless, A.; Hampton, K.K. Mechanisms of platelet aggregation by Streptococcus sanguis, a causative organism in infective endocarditis. Br. J. Haematol. 1993, 84, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Plummer, C.; Wu, H.; Kerrigan, S.W.; Meade, G.; Cox, D.; Ian Douglas, C.W. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 2005, 129, 101–109. [Google Scholar] [CrossRef]
- Yakovenko, O.; Nunez, J.; Bensing, B.; Yu, H.; Mount, J.; Zeng, J.; Hawkins, J.; Chen, X.; Sullam, P.M.; Thomas, W. Serine-Rich Repeat Adhesins Mediate Shear-Enhanced Streptococcal Binding to Platelets. Infect. Immun. 2018, 86, e00160-18. [Google Scholar] [CrossRef]
- Keane, C.; Petersen, H.J.; Tilley, D.; Haworth, J.; Cox, D.; Jenkinson, H.F.; Kerrigan, S.W. Multiple sites on Streptococcus gordonii surface protein PadA bind to platelet GPIIbIIIa. Thromb. Haemost. 2013, 110, 1278–1287. [Google Scholar] [CrossRef]
- Seo, H.S.; Xiong, Y.Q.; Sullam, P.M. Role of the serine-rich surface glycoprotein Srr1 of Streptococcus agalactiae in the pathogenesis of infective endocarditis. PLoS ONE 2013, 8, e64204. [Google Scholar] [CrossRef]
- McCormick, J.K.; Tripp, T.J.; Dunny, G.M.; Schlievert, P.M. Formation of vegetations during infective endocarditis excludes binding of bacterial-specific host antibodies to Enterococcus faecalis. J. Infect. Dis. 2002, 185, 994–997. [Google Scholar] [CrossRef]
- Nallapareddy, S.R.; Sillanpää, J.; Mitchell, J.; Singh, K.V.; Chowdhury, S.A.; Weinstock, G.M.; Sullam, P.M.; Murray, B.E. Conservation of Ebp-type pilus genes among Enterococci and demonstration of their role in adherence of Enterococcus faecalis to human platelets. Infect. Immun. 2011, 79, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Jamet, A.; Dervyn, R.; Lapaque, N.; Bugli, F.; Perez-Cortez, N.G.; Blottière, H.M.; Twizere, J.C.; Sanguinetti, M.; Posteraro, B.; Serror, P.; et al. The Enterococcus faecalis virulence factor ElrA interacts with the human Four-and-a-Half LIM Domains Protein 2. Sci. Rep. 2017, 7, 4581. [Google Scholar] [CrossRef] [PubMed]
- Johansson, D.; Rasmussen, M. Virulence factors in isolates of Enterococcus faecalis from infective endocarditis and from the normal flora. Microb. Pathog. 2013, 55, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Johansson, D.; Söbirk, S.K.; Mörgelin, M.; Shannon, O. Clinical isolates of Enterococcus faecalis aggregate human platelets. Microbes Infect. 2010, 12, 295–301. [Google Scholar] [CrossRef]
- Matos, R.C.; Lapaque, N.; Rigottier-Gois, L.; Debarbieux, L.; Meylheuc, T.; Gonzalez-Zorn, B.; Repoila, F.; Lopes, M.D.F.; Serror, P. Enterococcus faecalis Prophage Dynamics and Contributions to Pathogenic Traits. PLoS Genet. 2013, 9, e1003539. [Google Scholar] [CrossRef]
- Royer, G.; Roisin, L.; Demontant, V.; Lo, S.; Coutte, L.; Lim, P.; Pawlotsky, J.M.; Jacquier, H.; Lepeule, R.; Rodriguez, C.; et al. Microdiversity of Enterococcus faecalis isolates in cases of infective endocarditis: Selection of non-synonymous mutations and large deletions is associated with phenotypic modifications. Emerg. Microbes Infect. 2021, 10, 929–938. [Google Scholar] [CrossRef]
- Colomer-Winter, C.; Gaca, A.O.; Chuang-Smith, O.N.; Lemos, J.A.; Frank, K.L. Basal levels of (p)ppGpp differentially affect the pathogenesis of infective endocarditis in Enterococcus faecalis. Microbiology 2018, 164, 1254–1265. [Google Scholar] [CrossRef]
- Lepidi, H.; Houpikian, P.; Liang, Z.; Raoult, D. Cardiac valves in patients with Q fever endocarditis: Microbiological, molecular, and histologic studies. J. Infect. 2003, 187, 1097–1106. [Google Scholar] [CrossRef]
- Melenotte, C.; Epelboin, L.; Million, M.; Hubert, S.; Monsec, T.; Djossou, F.; Mège, J.L.; Habib, G.; Raoult, D. Acute Q Fever Endocarditis: A Paradigm Shift Following the Systematic Use of Transthoracic Echocardiography During Acute Q Fever. Clin. Infect. Dis. 2019, 69, 1987–1995. [Google Scholar] [CrossRef]
- Muretti, M.; Keiralla, A.; Jeffery, K.; Krasopoulos, G. Tropheryma whipplei endocarditis: An uncommon infection with potentially fatal consequences. J. Card. Surg. 2020, 35, 923–925. [Google Scholar] [CrossRef]
- García-Álvarez, L.; García-García, C.; Muñoz, P.; Fariñas-Álvarez, M.D.C.; Cuadra, M.G.; Fernández-Hidalgo, N.; García-Vázquez, E.; Moral-Escudero, E.; Alonso-Socas, M.D.M.; García-Rosado, D.; et al. Bartonella Endocarditis in Spain: Case Reports of 21 Cases. Pathogens 2022, 11, 561. [Google Scholar] [CrossRef] [PubMed]
- Hannachi, N.; Arregle, F.; Lepidi, H.; Baudoin, J.P.; Gouriet, F.; Martel, H.; Hubert, S.; Desnues, B.; Riberi, A.; Casalta, J.P.; et al. A Massive Number of Extracellular Tropheryma whipplei in Infective Endocarditis: A Case Report and Literature Review. Front. Immunol. 2022, 13, 900589. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Medvedev, A.E. Toll-Like Receptor Polymorphisms, Inflammatory and Infectious Diseases, Allergies, and Cancer. J. Interferon Cytokine Res. 2013, 33, 467–484. [Google Scholar] [CrossRef]
- Sharma, S.; Garg, I.; Ashraf, M.Z. TLR signalling and association of TLR polymorphism with cardiovascular diseases. Vasc. Pharmacol. 2016, 87, 30–37. [Google Scholar] [CrossRef]
- Golovkin, A.S.; Ponasenko, A.V.; Yuzhalin, A.E.; Salakhov, R.R.; Khutornaya, M.V.; Kutikhin, A.G.; Rutkovskaya, N.V.; Savostyanova, Y.Y.; Barbarash, L.S. An association between single nucleotide polymorphisms within TLR and TREM-1 genes and infective endocarditis. Cytokine 2015, 71, 16–21. [Google Scholar] [CrossRef]
- Bustamante, J.; Tamayo, E.; Flórez, S.; Telleria, J.J.; Bustamante, E.; López, J.; San Román, J.A.; Álvarez, F.J. Toll-Like Receptor 2 R753Q Polymorphisms Are Associated with an Increased Risk of Infective Endocarditis. Rev. Española Cardiol. (Engl. Ed.) 2011, 64, 1056–1059. [Google Scholar] [CrossRef]
- Liu, X.; Liu, H.; Luo, X.; Zhang, P.; Gao, Y.; Xie, S.; Xu, K.; Chang, J.; Ma, L. Strains of Group B streptococci from septic patients induce platelet activation via Toll-like Receptor 2. Clin. Exp. Pharmacol. Physiol. 2017, 44, 335–343. [Google Scholar] [CrossRef]
- Icli, A.; Tayyar, S.; Varol, E.; Aksoy, F.; Arslan, A.; Ersoy, I.; Akcay, S. Mean platelet volume is increased in infective endocarditis and decreases after treatment. Med. Princ. Pract. 2013, 22, 270–273. [Google Scholar] [CrossRef]
- Cho, S.Y.; Jeon, Y.L.; Kim, W.; Kim, W.S.; Lee, H.J.; Lee, W.I.; Park, T.S. Mean platelet volume and mean platelet volume/platelet count ratio in infective endocarditis. Platelets 2014, 25, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Gunebakmaz, O.; Kaya, M.G.; Kaya, E.G.; Ardic, I.; Yarlioglues, M.; Dogdu, O.; Kalay, N.; Akpek, M.; Sarli, B.; Ozdogru, I. Mean platelet volume predicts embolic complications and prognosis in infective endocarditis. Int. J. Infect. Dis. 2010, 14, e982–e985. [Google Scholar] [CrossRef] [PubMed]
- İleri, M.; Kanat, S.; Orhan, G.; Bayır, P.T.; Gürsoy, H.T.; Şahin, D.; Çiçek, G.; Elalmış, Ö.U.; Güray, Ü. Increased mean platelet volume in patients with infective endocarditis and embolic events. Cardiol. J. 2015, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Y.; He, X.; Ma, J.; Guo, W.; Dong, B.; Liang, W.; Wu, Y.; Owusu-Agyeman, M.; Xue, R.; et al. Mean platelet volume/platelet count ratio predicts long-term mortality in patients with infective endocarditis. Biomark. Med. 2020, 14, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Pafili, K.; Penlioglou, T.; Mikhailidis, D.P.; Papanas, N. Mean platelet volume and coronary artery disease. Curr. Opin. Cardiol. 2019, 34, 390–398. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Pu, J.; Jin, S.; Jia, L.; Li, X.; Liu, F.; Yang, Y. Mean platelet volume and coronary plaque vulnerability: An optical coherence tomography study in patients with non-ST-elevation acute coronary syndrome. BMC Cardiovasc. Disord. 2019, 19, 128. [Google Scholar] [CrossRef]
- Durack, D.T.; Beeson, P.B. Experimental bacterial endocarditis. I. Colonization of a sterile vegetation. Br. J. Exp. Pathol. 1972, 53, 44–49. [Google Scholar]
- Hannachi, N.; Lepidi, H.; Fontanini, A.; Takakura, T.; Bou-Khalil, J.; Gouriet, F.; Habib, G.; Raoult, D.; Camoin-Jau, L.; Baudoin, J.P. A Novel Approach for Detecting Unique Variations among Infectious Bacterial Species in Endocarditic Cardiac Valve Vegetation. Cells 2020, 9, 1899. [Google Scholar] [CrossRef]
- Liesenborghs, L.; Meyers, S.; Lox, M.; Criel, M.; Claes, J.; Peetermans, M.; Trenson, S.; Vande Velde, G.; Vanden Berghe, P.; Baatsen, P.; et al. Staphylococcus aureus endocarditis: Distinct mechanisms of bacterial adhesion to damaged and inflamed heart valves. Eur. Heart J. 2019, 40, 3248–3259. [Google Scholar] [CrossRef]
- Baudoin, J.P.; Camoin-Jau, L.; Prasanth, A.; Habib, G.; Lepidi, H.; Hannachi, N. Ultrastructure of a late-stage bacterial endocarditis valve vegetation. J. Thromb. Thrombolysis 2021, 51, 821–826. [Google Scholar] [CrossRef]
- Liesenborghs, L.; Meyers, S.; Vanassche, T.; Verhamme, P. Coagulation: At the heart of infective endocarditis. J. Thromb. Haemost. 2020, 18, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Sullam, P.M.; Frank, U.; Yeaman, M.R.; Tauber, M.G.; Bayer, A.S.; Chambers, H.F. Effect of thrombocytopenia on the early course of streptococcal endocarditis. J. Infect. Dis. 1993, 168, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Krijgsveld, J.; Zaat, S.A.; Meeldijk, J.; van Veelen, P.A.; Fang, G.; Poolman, B.; Brandt, E.; Ehlert, J.E.; Kuijpers, A.J.; Engbers, G.H.; et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J. Biol. Chem. 2000, 275, 20374–20381. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.S.; Ramos, M.D.; Menzies, B.E.; Yeaman, M.R.; Shen, A.J.; Cheung, A.L. Hyperproduction of alpha-toxin by Staphylococcus aureus results in paradoxically reduced virulence in experimental endocarditis: A host defense role for platelet microbicidal proteins. Infect. Immun. 1997, 65, 4652–4660. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Cody, M.J.; Franks, Z.; Tolley, N.D.; Kahr, W.H.; Lindemann, S.; Seizer, P.; Yost, C.C.; et al. Novel anti-bacterial activities of β-defensin 1 in human platelets: Suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. 2011, 7, e1002355. [Google Scholar] [CrossRef]
- Trier, D.A.; Gank, K.D.; Kupferwasser, D.; Yount, N.Y.; French, W.J.; Michelson, A.D.; Kupferwasser, L.I.; Xiong, Y.Q.; Bayer, A.S.; Yeaman, M.R. Platelet antistaphylococcal responses occur through P2X1 and P2Y12 receptor-induced activation and kinocidin release. Infect. Immun. 2008, 76, 5706–5713. [Google Scholar] [CrossRef]
- Hannachi, N.; Grac, L.; Baudoin, J.P.; Fournier, P.E.; Habib, G.; Camoin-Jau, L. Effect of antiplatelet agents on platelet antistaphylococcal capacity: An in vitro study. Int. J. Antimicrob. Agents 2020, 55, 105890. [Google Scholar] [CrossRef]
- Ezzeroug Ezzraimi, A.; Hannachi, N.; Mariotti, A.; Rolland, C.; Levasseur, A.; Baron, S.A.; Rolain, J.M.; Camoin-Jau, L. The Antibacterial Effect of Platelets on Escherichia coli Strains. Biomedicines 2022, 10, 1533. [Google Scholar] [CrossRef]
- Wu, T.; Yeaman, M.R.; Bayer, A.S. In vitro resistance to platelet microbicidal protein correlates with endocarditis source among bacteremic staphylococcal and streptococcal isolates. Antimicrob. Agents Chemother. 1994, 38, 729–732. [Google Scholar] [CrossRef]
- Kupferwasser, L.I.; Yeaman, M.R.; Shapiro, S.M.; Nast, C.C.; Bayer, A.S. In vitro susceptibility to thrombin-induced platelet microbicidal protein is associated with reduced disease progression and complication rates in experimental Staphylococcus aureus endocarditis: Microbiological, histopathologic, and echocardiographic analyses. Circulation 2002, 105, 746–752. [Google Scholar]
- Jung, C.J.; Yeh, C.Y.; Hsu, R.B.; Lee, C.M.; Shun, C.T.; Chia, J.S. Endocarditis pathogen promotes vegetation formation by inducing intravascular neutrophil extracellular traps through activated platelets. Circulation 2015, 131, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Stark, K. Platelet-neutrophil crosstalk and netosis. Hemasphere 2019, 3, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Hsu, R.B.; Ohniwa, R.L.; Chen, J.W.; Yuan, C.T.; Chia, J.S.; Jung, C.J. Neutrophil extracellular traps enhance Staphylococcus aureus vegetation formation through interaction with platelets in infective endocarditis. Thromb. Haemost. 2019, 119, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Meyers, S.; Crescente, M.; Verhamme, P.; Martinod, K. Staphylococcus aureus and Neutrophil Extracellular Traps: The Master Manipulator Meets Its Match in Immunothrombosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Mezger, M.; Nording, H.; Sauter, R.; Graf, T.; Heim, C.; von Bubnoff, N.; Ensminger, S.M.; Langer, H.F. Platelets and Immune Responses during Thromboinflammation. Front. Immunol. 2019, 10, 1731. [Google Scholar] [CrossRef]
- Tokarz-Deptuła, B.; Palma, J.; Baraniecki, Ł.; Stosik, M.; Kołacz, R.; Deptuła, W. What Function Do Platelets Play in Inflammation and Bacterial and Viral Infections? Front. Immunol. 2021, 12, 770436. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Nami, N.; Feci, L.; Napoliello, L.; Giordano, A.; Lorenzini, S.; Galeazzi, M.; Rubegni, P.; Fimiani, M. Crosstalk between platelets and PBMC: New evidence in wound healing. Platelets 2016, 27, 143–148. [Google Scholar] [CrossRef]
- Nording, H.; Langer, H.F. Complement links platelets to innate immunity. Semin. Immunol. 2018, 37, 43–52. [Google Scholar] [CrossRef]
- Veloso, T.R.; Que, Y.-A.; Chaouch, A.; Giddey, M.; Vouillamoz, J.; Rousson, V.; Moreillon, P.; Entenza, J.M. Prophylaxis of experimental endocarditis with antiplatelet and antithrombin agents: A role for long-term prevention of infective endocarditis in humans? J. Infect. Dis. 2015, 211, 72–79. [Google Scholar] [CrossRef]
- Veloso, T.R.; Oechslin, F.; Que, Y.A.; Moreillon, P.; Entenza, J.M.; Mancini, S. Aspirin plus ticlopidine prevented experimental endocarditis due to Enterococcus faecalis and Streptococcus gallolyticus. Pathog. Dis. 2015, 73, ftv060. [Google Scholar] [CrossRef] [PubMed]
- Anavekar, N.S.; Tleyjeh, I.M.; Anavekar, N.S.; Mirzoyev, Z.; Steckelberg, J.M.; Haddad, C.; Khandaker, M.H.; Wilson, W.R.; Chandrasekaran, K.; Baddour, L.M. Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin. Infect. Dis. 2007, 44, 1180–1186. [Google Scholar] [CrossRef]
- Anavekar, N.S.; Schultz, J.C.; De Sa, D.D.C.; Thomas, J.M.; Lahr, B.D.; Tleyjeh, I.M.; Steckelberg, J.M.; Wilson, W.R.; Baddour, L.M. Modifiers of symptomatic embolic risk in infective endocarditis. Mayo Clin. Proc. 2011, 86, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; Tam, J.; Dumesnil, J.G.; Cujec, B.; Sanfilippo, A.J.; Jue, J.; Turek, M.; Robinson, T.; Williams, K. Effect of long-term aspirin use on embolic events in infective endocarditis. Clin. Infect. Dis. 2008, 46, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Pepin, J.; Tremblay, V.; Bechard, D.; Rodier, F.; Walker, C.; Dufresne, D.; Lafontaine, A.; Li, N.; Lacroix, C.; Lanthier, L. Chronic antiplatelet therapy and mortality among patients with infective endocarditis. Clin. Microbiol. Infect. 2009, 15, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Snygg-Martin, U.; Rasmussen, R.V.; Hassager, C.; Bruun, N.E.; Andersson, R.; Olaison, L. The relationship between cerebrovascular complications and previously established use of antiplatelet therapy in leftsided infective endocarditis. Scand. J. Infect. Dis. 2011, 43, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Eisen, D.P.; Corey, G.R.; McBryde, E.S.; Fowler, V.G.; Miro, J.M.; Cabell, C.H.; Street, A.C.; Paiva, M.G.; Ionac, A.; Tan, R.S.; et al. Reduced valve replacement surgery and complication rate in Staphylococcus aureus endocarditis patients receiving acetyl-salicylic acid. J. Infect. 2009, 58, 332–338. [Google Scholar] [CrossRef]
- Chan, K.-L.; Dumesnil, J.G.; Cujec, B.; Sanfilippo, A.J.; Jue, J.; Turek, M.A.; Robinson, T.; Moher, D. A randomized trial of aspirin on the risk of embolic events in patients with infective endocarditis. J. Am. Coll. Cardiol. 2003, 42, 775–780. [Google Scholar] [CrossRef]
- Hannachi, N.; Ogé-Ganaye, E.; Baudoin, J.P.; Fontanini, A.; Bernot, D.; Habib, G.; Camoin-Jau, L. Antiplatelet Agents Have a Distinct Efficacy on Platelet Aggregation Induced by Infectious Bacteria. Front. Pharmacol. 2020, 11, 863. [Google Scholar] [CrossRef]
- Caffrey, A.R.; Appaneal, H.J.; LaPlante, K.L.; Lopes, V.V.; Ulloa, E.R.; Nizet, V.; Sakoulas, G. Impact of Clopidogrel on Clinical Outcomes in Patients with Staphylococcus aureus Bacteremia: A National Retrospective Cohort Study. Antimicrob. Agents Chemother. 2022, 66, e0211721. [Google Scholar] [CrossRef]
- Storey, R.F.; James, S.K.; Siegbahn, A.; Varenhorst, C.; Held, C.; Ycas, J.; Husted, S.E.; Cannon, C.P.; Becker, R.C.; Steg, P.G.; et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets 2014, 25, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Lupu, L.; Shepshelovich, D.; Banai, S.; Hershkoviz, R.; Isakov, O. Effect of Ticagrelor on Reducing the Risk of Gram-Positive Infections in Patients with Acute Coronary Syndrome. Am. J. Cardiol. 2020, 130, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lin, H.W.; Lee, N.Y.; Lin, S.H.; Li, Y.H. Risk of infectious events in acute myocardial infarction patients treated with ticagrelor or clopidogrel. Eur. J. Intern. Med. 2021, 85, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Butt, J.H.; Fosbøl, E.L.; Gerds, T.A.; Iversen, K.; Bundgaard, H.; Bruun, N.E.; Larsen, A.R.; Petersen, A.; Andersen, P.S.; Skov, R.L.; et al. Ticagrelor and the risk of Staphylococcus aureus bacteraemia and other infections. Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, P.; Musumeci, L.; Jacques, N.; Servais, L.; Goffin, E.; Pirotte, B.; Oury, C. Antibacterial Activity of Ticagrelor in Conventional Antiplatelet Dosages Against Antibiotic-Resistant Gram-Positive Bacteria. JAMA Cardiol. 2019, 4, 596–599. [Google Scholar] [CrossRef]
- Meyer, O.O.; Thill, C.J. Penicillin and dicumarol, and penicillin in the treatment of subacute bacterial endocarditis. Proc. Annu. Meet. Cent. Soc. Clin. Res. (U.S.) 1945, 18, 31. [Google Scholar] [PubMed]
- Priest, W.S.; Smith, J.M.; McGee, C.J. The effect of anticoagulants on the penicillin therapy and the pathologic lesions of subacute bacterial endocarditis. N. Engl. J. Med. 1946, 235, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Loewe, L.; Rosenblatt, P.; Greene, H.J. Combined Penicillin and Heparin Therapy of Subacute Bacterial Endocarditis. Bull. N. Y. Acad. Med. 1946, 22, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Tornos, P.; Almirante, B.; Mirabet, S.; Permanyer, G.; Pahissa, A.; Soler-Soler, J. Infective endocarditis due to Staphylococcus aureus: Deleterious effect of anticoagulant therapy. Arch. Intern. Med. 1999, 159, 473–475. [Google Scholar] [CrossRef]
- Vanassche, T.; Verhaegen, J.; Peetermans, W.E.; Hoylaerts, M.F.; Verhamme, P. Dabigatran inhibits Staphylococcus aureus coagulase activity. J. Clin. Microbiol. 2010, 48, 4248–4250. [Google Scholar] [CrossRef]
- Vanassche, T.; Verhaegen, J.; Peetermans, W.E.; Van Ryn, J.; Cheng, A.; Schneewind, O.; Hoylaerts, M.F.; Verhamme, P. Inhibition of staphylothrombin by dabigatran reduces Staphylococcus aureus virulence. J. Thromb. Haemost. 2011, 9, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Lerche, C.J.; Christophersen, L.J.; Goetze, J.P.; Nielsen, P.R.; Thomsen, K.; Enevold, C.; Høiby, N.; Jensen, P.Ø.; Bundgaard, H.; Moser, C. Adjunctive dabigatran therapy improves outcome of experimental left-sided Staphylococcus aureus endocarditis. PLoS ONE 2019, 14, e0215333. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.F.; Chu, H.; Ou, S.M.; Li, S.Y.; Chen, Y.T.; Shih, C.J.; Tsai, L.W. Effect of statin therapy on mortality in patients with infective endocarditis. Am. J. Cardiol. 2014, 114, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [PubMed]
- Nenna, A.; Nappi, F.; Lusini, M.; Satriano, U.M.; Schilirò, D.; Spadaccio, C.; Chello, M. Effect of Statins on Platelet Activation and Function: From Molecular Pathways to Clinical Effects. Biomed. Res. Int. 2021, 2021, 6661847. [Google Scholar] [CrossRef]
- Hannachi, N.; Fournier, P.E.; Martel, H.; Habib, G.; Camoin-Jau, L. Statins potentiate the antibacterial effect of platelets on Staphylococcus aureus. Platelets 2021, 32, 671–676. [Google Scholar] [CrossRef]
- Yu, S.Y.; Li, H.L.; Tse, Y.K.; Li, X.; Ren, Q.W.; Wu, M.Z.; Wong, P.F.; Tse, H.F.; Lip, G.Y.H.; Yiu, K.H. Pre-admission and In-Hospital Statin Use is Associated with Reduced Short Term Mortality in Infective Endocarditis. Mayo. Clin. Proc. 2022, 98, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Munnix, I.C.A.; Van Oerle, R.; Verhezen, P.; Kuijper, P.; Hackeng, C.M.; Hopman-Kerkhoff, H.I.J.; Hudig, F.; Van De Kerkhof, D.; Leyte, A.; De Maat, M.P.M.; et al. Harmonizing light transmission aggregometry in the Netherlands by implementation of the SSC-ISTH guideline. Platelets 2021, 32, 516–523. [Google Scholar] [CrossRef]
- Clawson, C.C.; White, J.G. Platelet Interaction with Bacteria. Am. J. Pathol. 1971, 65, 367–380. [Google Scholar]
- Montague, S.J.; Lim, Y.J.; Lee, W.M.; Gardiner, E.E. Imaging Platelet Processes and Function-Current and Emerging Approaches for Imaging in vitro and in vivo. Front. Immunol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Arbesu, I.; Bucsaiova, M.; Fischer, M.B.; Mannhalter, C. Platelet-borne complement proteins and their role in platelet–bacteria interactions. J. Thromb. Haemost. 2016, 14, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Arregle, F.; Gouriet, F.; Amphoux, B.; Edouard, S.; Chaudet, H.; Casalta, J.P.; Habib, G.; Fournier, P.E.; Raoult, D. Western Immunoblotting for the Diagnosis of Enterococcus faecalis and Streptococcus gallolyticus Infective Endocarditis. Front. Cell Infect. Microbiol. 2019, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Gnatenko, D.V.; Dunn, J.J.; McCorkle, S.R.; Weissmann, D.; Perrotta, P.L.; Bahou, W.F. Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood 2003, 101, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Clawson, C.C. Platelet Interaction with Bacteria. Am. J. Pathol. 1973, 70, 449–472. [Google Scholar]
- Chen, D.; Uhl, C.B.; Bryant, S.C.; Krumwiede, M.; Barness, R.L.; Olson, M.C.; Gossman, S.C.; Damgard, S.E.; Gamb, S.I.; Cummins, L.A.; et al. Diagnostic laboratory standardization and validation of platelet transmission electron microscopy. Platelets 2018, 29, 574–582. [Google Scholar] [CrossRef]
- Ezzeroug Ezzraimi, A.; Baudoin, J.P.; Mariotti, A.; Camoin-Jau, L. Microscopic Description of Platelet Aggregates Induced by Escherichia coli Strains. Cells 2022, 11, 3495. [Google Scholar] [CrossRef]
- Prudent, E.; Lepidi, H.; Angelakis, E.; Raoult, D. Fluorescence In Situ Hybridization (FISH) and Peptide Nucleic Acid Probe-Based FISH for Diagnosis of Q Fever Endocarditis and Vascular Infections. J. Clin. Microbiol. 2018, 56, e00542-18. [Google Scholar] [CrossRef]
- Ge, X.; Kitten, T.; Chen, Z.; Lee, S.P.; Munro, C.L.; Xu, P. Identification of Streptococcus sanguinis Genes Required for Biofilm Formation and Examination of Their Role in Endocarditis Virulence. Infect. Immun. 2008, 76, 2551–2559. [Google Scholar] [CrossRef]
- Xiong, Y.Q.; Willard, J.; Yeaman, M.R.; Cheung, A.L.; Bayer, A.S. Regulation of Staphylococcus aureus α-Toxin Gene (hla) Expression by agr, sarA and sae In Vitro and in Experimental Infective Endocarditis. J. Infect. Dis. 2006, 194, 1267–1275. [Google Scholar] [CrossRef]
- Dall, L.; Miller, T.; Herndon, B.; Diez, I.; Dew, M. Platelet depletion and severity of streptococcal endocarditis. Can. J. Infect. Dis. 1998, 9, 359–366. [Google Scholar] [CrossRef]
- Nicolau, D.P.; Tessier, P.R.; Nightingale, C.H.; Quintiliani, R. Influence of adjunctive ticlopidine on the treatment of experimental Staphylococcus aureus endocarditis. Int. J. Antimicrob. Agents 1998, 9, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Habib, G.; Lancellotti, P.; Antunes, M.J.; Bongiorni, M.G.; Casalta, J.-P.; Del Zotti, F.; Dulgheru, R.; El Khoury, G.; Erba, P.A.; Iung, B.; et al. 2015 ESC Guidelines for the management of infective endocarditis: The Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur. Heart J. 2015, 36, 3075–3128. [Google Scholar] [CrossRef] [PubMed]


| Platelet aggregation, platelet–leukocyte aggregation, activation of coagulation | [75] |
| Secretion of bactericidal substances contained in their alpha granules and mechanical removal of bacteria | [63,64,65,76] |
| Activation of NETosis | [71,72,73,74,77] |
| Synthesis by platelets of immune mediators for the activation of T and B lymphocytes, macrophages, and dendritic cells | [78] |
| Activation of complement system | [75] |
| Activation of endothelium and extravasation | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braï, M.A.; Hannachi, N.; El Gueddari, N.; Baudoin, J.-P.; Dahmani, A.; Lepidi, H.; Habib, G.; Camoin-Jau, L. The Role of Platelets in Infective Endocarditis. Int. J. Mol. Sci. 2023, 24, 7540. https://doi.org/10.3390/ijms24087540
Braï MA, Hannachi N, El Gueddari N, Baudoin J-P, Dahmani A, Lepidi H, Habib G, Camoin-Jau L. The Role of Platelets in Infective Endocarditis. International Journal of Molecular Sciences. 2023; 24(8):7540. https://doi.org/10.3390/ijms24087540
Chicago/Turabian StyleBraï, Mustapha Abdeljalil, Nadji Hannachi, Nabila El Gueddari, Jean-Pierre Baudoin, Abderrhamane Dahmani, Hubert Lepidi, Gilbert Habib, and Laurence Camoin-Jau. 2023. "The Role of Platelets in Infective Endocarditis" International Journal of Molecular Sciences 24, no. 8: 7540. https://doi.org/10.3390/ijms24087540
APA StyleBraï, M. A., Hannachi, N., El Gueddari, N., Baudoin, J. -P., Dahmani, A., Lepidi, H., Habib, G., & Camoin-Jau, L. (2023). The Role of Platelets in Infective Endocarditis. International Journal of Molecular Sciences, 24(8), 7540. https://doi.org/10.3390/ijms24087540