

Mutated Flt3Lg Provides Reduced Flt3 Recycling Compared to Wild-Type Flt3Lg and Retains the Specificity of Flt3Lg-Based CAR T-Cell Targeting in AML Models

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Recombinant Flt3Lg-L27P Did Not Disrupt Wild-Type Flt3Lg Bioactivity
2.2. Low Doses of Recombinant Flt3Lg-L27P Did Not Stimulate Flt3 Downregulation, While Flt3Lg Did
2.3. Generating Flt3m-CAR T-Cells
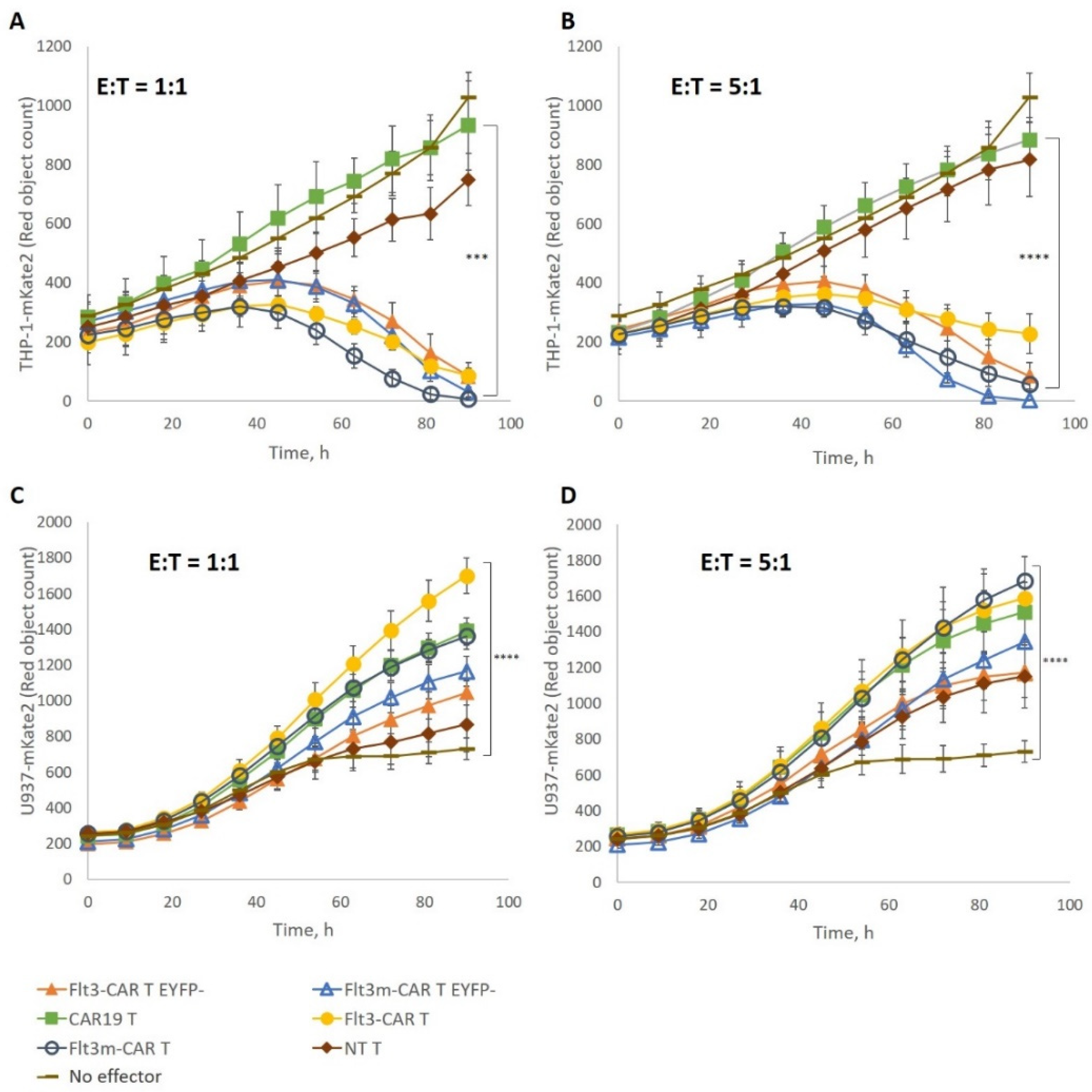
2.4. Flt3m-CAR T-Cells Showed Cytotoxicity Similar to That of Flt3-CAR T-Cells
2.5. Recombinant Flt3Lg-L27P Did Not Interfere with Flt3m-CAR T-Cell-Mediated Killing
2.6. Excess of Flt3m-CAR Jurkat Cells Could Not Interfere in Flt3m-CAR T-Cell-Mediated Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Flt3Lg-L27P Cloning and Purification
4.2. CAR Lentiviruses Generation
4.3. Cell Culture
4.4. Flt3m-CAR T-Cell and CAR Jurkat Cell Generation
4.5. Western-Blotting
4.6. Flow Cytometry
4.7. CAR T-Cell and CAR Jurkat Cell Cytotoxicity Assay
4.8. Cytokine Secretion
4.9. Proliferation Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Marvin-Peek, J.; Savani, B.N.; Olalekan, O.O.; Dholaria, B. Challenges and Advances in Chimeric Antigen Receptor Therapy for Acute Myeloid Leukemia. Cancers 2022, 14, 497. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1407222 (accessed on 11 January 2022).
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. The Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Munshi, N.C.; Larry, D.; Anderson, J.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Davey, A.S.; Call, M.E.; Call, M.J. The Influence of Chimeric Antigen Receptor Structural Domains on Clinical Outcomes and Associated Toxicities. Cancers 2020, 13, 38. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Nieba, L.; Honegger, A.; Krebber, C.; Plückthun, A. Disrupting the Hydrophobic Patches at the Antibody Variable/Constant Domain Interface: Improved in Vivo Folding and Physical Characterization of an Engineered ScFv Fragment. Protein Eng. Des. Sel. 1997, 10, 435–444. [Google Scholar] [CrossRef]
- Gil, D.; Schrum, A.G. Strategies to Stabilize Compact Folding and Minimize Aggregation of Antibody-Based Fragments. Adv. Biosci. Biotechnol. 2013, 4, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Salzer, B.; Schueller, C.M.; Zajc, C.U.; Peters, T.; Schoeber, M.A.; Kovacic, B.; Buri, M.C.; Lobner, E.; Dushek, O.; Huppa, J.B.; et al. Engineering AvidCARs for Combinatorial Antigen Recognition and Reversible Control of CAR Function. Nat. Commun. 2020, 11, 4166. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-Tuning the CAR Spacer Improves T-Cell Potency. OncoImmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Maiorova, V.; Mollaev, M.D.; Vikhreva, P.; Kulakovskaya, E.; Pershin, D.; Chudakov, D.M.; Kibardin, A.; Maschan, M.A.; Larin, S. Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines. Vaccines 2021, 9, 1238. [Google Scholar] [CrossRef]
- Graddis, T.J.; Brasel, K.; Friend, D.; Srinivasan, S.; Wee, S.; Lyman, S.D.; March, C.J.; McGrew, J.T. Structure-Function Analysis of FLT3 Ligand-FLT3 Receptor Interactions Using a Rapid Functional Screen. J. Biol. Chem. 1998, 273, 17626–17633. [Google Scholar] [CrossRef]
- Pannecoucke, E.; Raes, L.; Savvides, S.N. Engineering and Crystal Structure of a Monomeric FLT3 Ligand Variant. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2021, 77, 121–127. [Google Scholar] [CrossRef]
- Oveland, E.; Gjertsen, B.T.; Wergeland, L.; Selheim, F.; Fladmark, K.E.; Hovland, R. Ligand-Induced Flt3-Downregulation Modulates Cell Death Associated Proteins and Enhances Chemosensitivity to Idarubicin in THP-1 Acute Myeloid Leukemia Cells. Leuk. Res. 2009, 33, 276–287. [Google Scholar] [CrossRef]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T Cell Activation by Antibody-Like Immunoreceptors: Increase in Affinity of the Single-Chain Fragment Domain above Threshold Does Not Increase T Cell Activation against Antigen-Positive Target Cells but Decreases Selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.F.; Shannon, M.F.; Hercus, T.; Nicola, N.A.; Cambareri, B.; Dottore, M.; Layton, M.J.; Eglinton, L.; Vadas, M.A. Residue 21 of Human Granulocyte-Macrophage Colony-Stimulating Factor Is Critical for Biological Activity and for High but Not Low Affinity Binding. EMBO J. 1992, 11, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, A.; Saito, S.; Narimatsu, S.; Nakano, S.; Nagai, M.; Ohnota, H.; Inada, Y.; Morokawa, H.; Nakashima, I.; Morita, D.; et al. Mutated GM-CSF-Based CAR-T Cells Targeting CD116/CD131 Complexes Exhibit Enhanced Anti-Tumor Effects against Acute Myeloid Leukaemia. Clin. Transl. Immunol. 2021, 10, e1282. [Google Scholar] [CrossRef] [PubMed]
- Zoine, J.T.; Prince, C.; Story, J.Y.; Branella, G.M.; Lytle, A.M.; Fedanov, A.; Alexander, J.S.; Porter, C.C.; Doering, C.B.; Spencer, H.T.; et al. Thrombopoietin-Based CAR-T Cells Demonstrate in Vitro and in Vivo Cytotoxicity to MPL Positive Acute Myelogenous Leukemia and Hematopoietic Stem Cells. Gene Ther. 2022, 29, 1–12. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem Cell Factor Receptor/c-Kit: From Basic Science to Clinical Implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, P.; Chevallier, P.; Knapper, S.; Collin, M. FLT3 Ligand in Acute Myeloid Leukemia: A Simple Test with Deep Implications. Leuk. Lymphoma 2021, 62, 264–270. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiorova, V.; Mollaev, M.D.; Vikhreva, P.; Chudakov, D.M.; Kibardin, A.; Maschan, M.A.; Larin, S. Mutated Flt3Lg Provides Reduced Flt3 Recycling Compared to Wild-Type Flt3Lg and Retains the Specificity of Flt3Lg-Based CAR T-Cell Targeting in AML Models. Int. J. Mol. Sci. 2023, 24, 7626. https://doi.org/10.3390/ijms24087626
Maiorova V, Mollaev MD, Vikhreva P, Chudakov DM, Kibardin A, Maschan MA, Larin S. Mutated Flt3Lg Provides Reduced Flt3 Recycling Compared to Wild-Type Flt3Lg and Retains the Specificity of Flt3Lg-Based CAR T-Cell Targeting in AML Models. International Journal of Molecular Sciences. 2023; 24(8):7626. https://doi.org/10.3390/ijms24087626
Chicago/Turabian StyleMaiorova, Varvara, Murad D. Mollaev, Polina Vikhreva, Dmitriy M. Chudakov, Alexey Kibardin, Michael A. Maschan, and Sergey Larin. 2023. "Mutated Flt3Lg Provides Reduced Flt3 Recycling Compared to Wild-Type Flt3Lg and Retains the Specificity of Flt3Lg-Based CAR T-Cell Targeting in AML Models" International Journal of Molecular Sciences 24, no. 8: 7626. https://doi.org/10.3390/ijms24087626
APA StyleMaiorova, V., Mollaev, M. D., Vikhreva, P., Chudakov, D. M., Kibardin, A., Maschan, M. A., & Larin, S. (2023). Mutated Flt3Lg Provides Reduced Flt3 Recycling Compared to Wild-Type Flt3Lg and Retains the Specificity of Flt3Lg-Based CAR T-Cell Targeting in AML Models. International Journal of Molecular Sciences, 24(8), 7626. https://doi.org/10.3390/ijms24087626