
Inhibition of ITK Signaling Causes Amelioration in Sepsis-Associated Neuroinflammation and Depression-like State in Mice
 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sepsis Causes Upregulation of ITK in the Periphery and CNS
2.2. Sepsis-Induced Elevation in Th17 Cell-Related Signaling Is Attenuated by ITK Inhibition
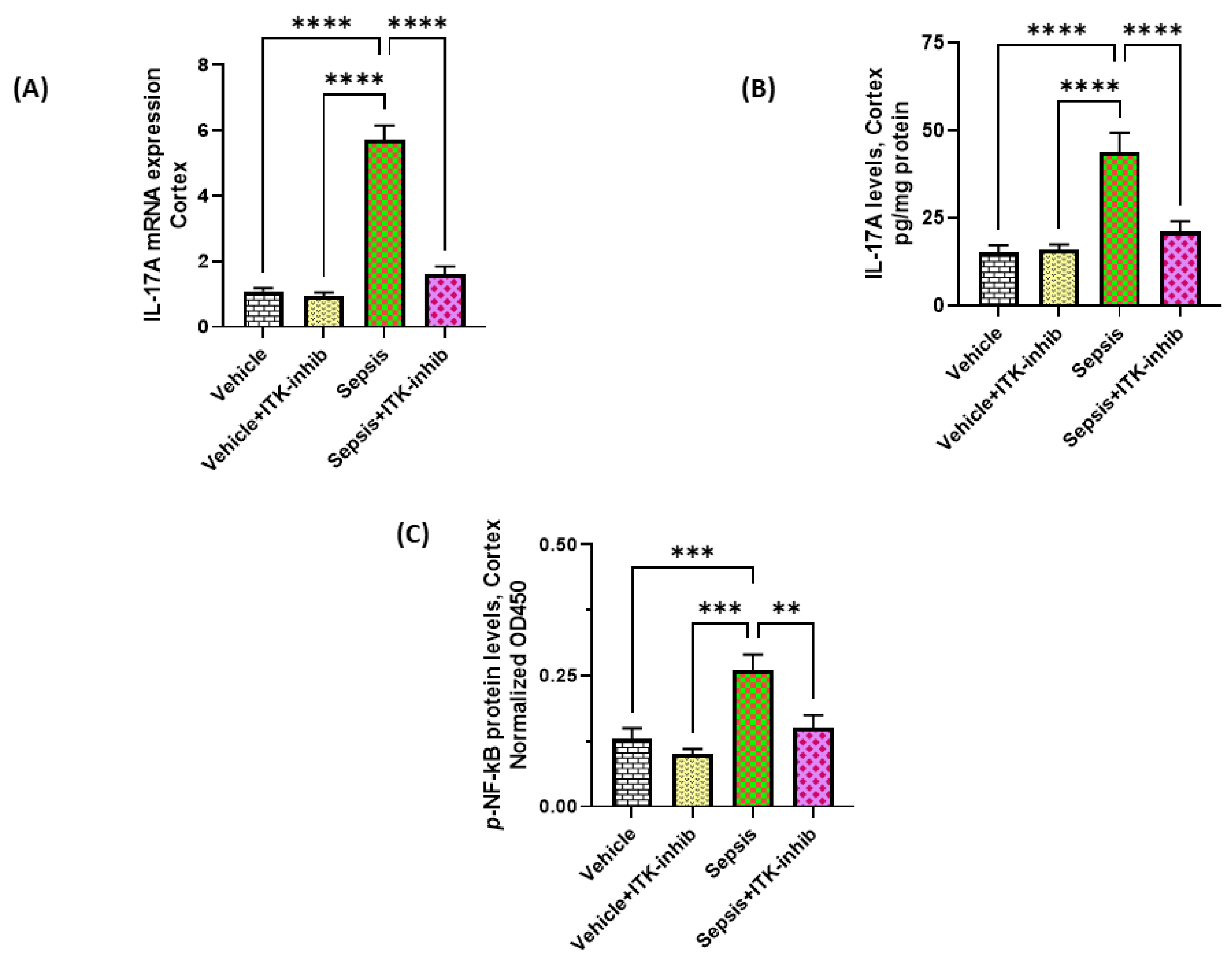
2.3. Sepsis-Induced Elevation in IL-17A Signaling Is Attenuated by ITK Inhibition
2.4. Sepsis-Induced Oxidative Neuroinflammation Is Attenuated by ITK Inhibition
2.5. Treg Cell-Related Signaling Is Further Elevated by ITK Inhibition in Sepsis Survivor Mice
2.6. Sepsis-Mediated Decrease in Antioxidant Transcription Factor Nrf2 Is Restored by ITK Inhibition in the CNS
2.7. Sepsis-Induced Depression-like Behavior Is Ameliorated by ITK Inhibition
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of Sepsis and Drug Administration
4.3. Tail Suspension Method
4.4. Sucrose Preference Test
4.5. Marble Bury Behavior
4.6. Measurement of Myeloperoxidase Activity in CNS
4.7. Nrf2 Trans-Activation ELISA Assay in the CNS
4.8. Real-Time PCR
4.9. Preparation of Samples for Biochemical Measurements
4.10. Estimation of ITK, p-NFkB, and p-STAT3 Levels in the Cortex
4.11. Estimation of Lipid Peroxides in Cortex
4.12. Estimation of Cytokines in Cortex/Serum
4.13. Flow Cytometry
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dellinger, R.P.; Levy, M.M.; Rhodes, A.; Annane, D.; Gerlach, H.; Opal, S.M.; Sevransky, J.E.; Sprung, C.L.; Douglas, I.S.; Jaeschke, R.; et al. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013, 41, 580–637. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Artigas, A.; Phillips, G.S.; Rhodes, A.; Beale, R.; Osborn, T.; Vincent, J.-L.; Townsend, S.; Lemeshow, S.; Dellinger, R.P. Outcomes of the Surviving Sepsis Campaign in intensive care units in the USA and Europe: A prospective cohort study. Lancet Infect. Dis. 2012, 12, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- De La Rica, A.S.; Gilsanz, F.; Maseda, E. Epidemiologic trends of sepsis in western countries. Ann. Transl. Med. 2016, 4, 325. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, R.; Jiang, L.; Li, S.; Xue, Y. Basic research and clinical progress of sepsis-associated encephalopathy. J. Intensiv. Med. 2021, 1, 90–95. [Google Scholar] [CrossRef]
- Kaukonen, K.M.; Bailey, M.; Suzuki, S.; Pilcher, D.; Bellomo, R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA 2014, 311, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Yao, R.-Q.; Zhang, H.; Feng, Y.-W.; Yao, Y.-M. Sepsis-associated encephalopathy: A vicious cycle of immunosuppression. J. Neuroinflammation 2020, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Adam, N.; Kandelman, S.; Mantz, J.; Chrétien, F.; Sharshar, T. Sepsis-induced brain dysfunction. Expert Rev. Anti-infective Ther. 2013, 11, 211–221. [Google Scholar] [CrossRef]
- Gallagher, M.P.; Conley, J.M.; Vangala, P.; Garber, M.; Reboldi, A.; Berg, L.J. Hierarchy of signaling thresholds downstream of the T cell receptor and the Tec kinase ITK. Proc. Natl. Acad. Sci. USA 2021, 118, e2025825118. [Google Scholar] [CrossRef]
- Lechner, K.S.; Neurath, M.F.; Weigmann, B. Role of the IL-2 inducible tyrosine kinase ITK and its inhibitors in disease pathogenesis. J. Mol. Med. 2020, 98, 1385–1395. [Google Scholar] [CrossRef]
- Mammadli, M.; Harris, R.; Suo, L.; May, A.; Gentile, T.; Waickman, A.T.; Bah, A.; August, A.; Nurmemmedov, E.; Karimi, M. Interleu-kin-2-inducible T-cell kinase (Itk) signaling regulates potent noncanonical regulatory T cells. Clin. Transl. Med. 2021, 11, e625. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Jeong, A.-R.; Kannan, A.K.; Huang, L.; August, A. IL-2–Inducible T Cell Kinase Tunes T Regulatory Cell Development and Is Required for Suppressive Function. J. Immunol. 2014, 193, 2267–2272. [Google Scholar] [CrossRef]
- Weeks, S.; Harris, R.; Karimi, M. Targeting ITK signaling for T cell-mediated diseases. iScience 2021, 24, 102842. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, A.H.; Schwartzberg, P.L.; Joseph, R.E.; Berg, L.J. T-Cell Signaling Regulated by the Tec Family Kinase, Itk. Cold Spring Harb. Perspect. Biol. 2010, 2, a002287. [Google Scholar] [CrossRef] [PubMed]
- Berg, L.J.; Finkelstein, L.D.; Lucas, J.A.; Schwartzberg, P.L.; Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S.; Thomas, S.M.; Brugge, J.S.; Veillette, A.; et al. Tec family kinases in t lymphocyte development and function. Annu. Rev. Immunol. 2005, 23, 549–600. [Google Scholar] [CrossRef]
- Gomez-Rodriguez, J.; Kraus, Z.J.; Schwartzberg, P.L. Tec family kinases Itk and Rlk / Txk in T lymphocytes: Cross-regulation of cytokine production and T-cell fates. FEBS J. 2011, 278, 1980–1989. [Google Scholar] [CrossRef]
- Kannan, A.K.; Kim, D.-G.; August, A.; Bynoe, M.S. Itk Signals Promote Neuroinflammation by Regulating CD4+T-Cell Activation and Trafficking. J. Neurosci. 2015, 35, 221–233. [Google Scholar] [CrossRef]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Ibrahim, K.E.; Alqahtani, F.; Sobeai, H.M.A.; Alotaibi, M.R. Inhibition of interleukin-2-inducible T-cell kinase causes reduction in imiquimod-induced psoriasiform inflammation through reduction of Th17 cells and en-hancement of Treg cells in mice. Biochimie 2020, 179, 146–156. [Google Scholar] [CrossRef]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Reyes, M.; Filbin, M.R.; Bhattacharyya, R.P.; Billman, K.; Eisenhaure, T.; Hung, D.T.; Levy, B.D.; Baron, R.M.; Blainey, P.C.; Goldberg, M.B.; et al. An immune-cell signature of bacterial sepsis. Nat. Med. 2020, 26, 333–340. [Google Scholar] [CrossRef]
- Anderson, S.T.; Commins, S.; Moynagh, P.N.; Coogan, A.N. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain Behav. Immun. 2015, 43, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, R.; Mader, S.; Valdés-Ferrer, S.I. Systemic inflammation and the brain: Novel roles of genetic, molecular, and envi-ronmental cues as drivers of neurodegeneration. Front. Cell. Neurosci. 2015, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-F.; Cao, K.; Jiang, J.-P.; Guan, W.-X.; Du, J.-F. Neutrophil dysregulation during sepsis: An overview and update. J. Cell. Mol. Med. 2017, 21, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Comim, C.M.; Vilela, M.C.; Constantino, L.; Petronilho, F.; Vuolo, F.; Lacerda-Queiroz, N.; Rodrigues, D.H.; Da Rocha, J.L.; Teixeira, A.L.; de Quevedo, J.L.; et al. Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intensiv. Care Med. 2011, 37, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Mei, X.-L.; Zhao, Y.-N. Sepsis and Cerebral Dysfunction: BBB Damage, Neuroinflammation, Oxidative Stress, Apoptosis and Autophagy as Key Mediators and the Potential Therapeutic Approaches. Neurotox. Res. 2021, 39, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Andrades, M.; Morina, A.; Spasić, S.; Spasojević, I. Bench-to-bedside review: Sepsis—From the redox point of view. Crit. Care 2011, 15, 230. [Google Scholar] [CrossRef]
- Delano, M.J.; Ward, P.A. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Invest. 2016, 126, 23–31. [Google Scholar] [CrossRef]
- Kim, K.S.; Jekarl, D.W.; Yoo, J.; Lee, S.; Kim, M.; Kim, Y. Immune gene expression networks in sepsis: A network biology approach. PLoS ONE 2021, 16, e0247669. [Google Scholar] [CrossRef]
- Catorce, M.N.; Gevorkian, G. Evaluation of Anti-inflammatory Nutraceuticals in LPS-induced Mouse Neuroinflammation Model: An Update. Curr. Neuropharmacol. 2020, 18, 636–654. [Google Scholar] [CrossRef]
- Xu, X.-E.; Liu, L.; Wang, Y.-C.; Wang, C.-T.; Zheng, Q.; Liu, Q.-X.; Li, Z.-F.; Bai, X.-J.; Liu, X.-H. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav. Immun. 2019, 80, 859–870. [Google Scholar] [CrossRef]
- Elmore, J.P.; McGee, M.C.; Nidetz, N.F.; Anannya, O.; Huang, W.; August, A. Tuning T helper cell differentiation by ITK. Biochem. Soc. Trans. 2020, 48, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Neuroimmune communication. Nat. Neurosci. 2017, 20, 127. [CrossRef] [PubMed]
- Zarbato, G.F.; de Souza Goldim, M.P.; Giustina, A.D.; Danielski, L.G.; Mathias, K.; Florentino, D.; de Oliveira Junior, A.N.; da Rosa, N.; Laurentino, A.O.; Trombetta, T.; et al. Dimethyl Fumarate Limits Neu-roinflammation and Oxidative Stress and Improves Cognitive Impairment After Polymicrobial Sepsis. Neurotox. Res. 2018, 34, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa-Ishii, S.; Inaba, M.; Umegaki, H.; Unno, K.; Wakabayashi, K.; Shimada, A. Endotoxemia-induced cytokine-mediated re-sponses of hippocampal astrocytes transmitted by cells of the brain-immune interface. Sci. Rep. 2016, 6, 25457. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wei, B.; Li, L.; Wang, B.; Sun, M. Th17 cells and inflammation in neurological disorders: Possible mechanisms of action. Front. Immunol. 2022, 13, 932152. [Google Scholar] [CrossRef]
- Shimada, A.; Hasegawa-Ishii, S. Histological Architecture Underlying Brain–Immune Cell–Cell Interactions and the Cerebral Response to Systemic Inflammation. Front. Immunol. 2017, 8, 17. [Google Scholar] [CrossRef]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Fardan, A.S.; El-Sherbeeny, A.M.; Ibrahim, K.E.; Attia, S.M. IL-17A causes depression-like symptoms via NFκB and p38MAPK signaling pathways in mice: Implications for psoriasis associated depression. Cytokine 2017, 97, 14–24. [Google Scholar] [CrossRef]
- Guo, K.; Zhang, X. Cytokines that Modulate the Differentiation of Th17 Cells in Autoimmune Uveitis. J. Immunol. Res. 2021, 2021, 6693542. [Google Scholar] [CrossRef]
- Saito, M.; Fujinami, Y.; Ono, Y.; Ohyama, S.; Fujioka, K.; Yamashita, K.; Inoue, S.; Kotani, J. Infiltrated regulatory T cells and Th2 cells in the brain contribute to attenuation of sepsis-associated encephalopathy and alleviation of mental impairments in mice with polymicrobial sepsis. Brain Behav. Immun. 2021, 92, 25–38. [Google Scholar] [CrossRef]
- Ye, B.; Tao, T.; Zhao, A.; Wen, L.; He, X.; Liu, Y.; Fu, Q.; Mi, W.; Lou, J. Blockade of IL-17A/IL-17R Pathway Protected Mice from Sep-sis-Associated Encephalopathy by Inhibition of Microglia Activation. Mediat. Inflamm. 2019, 2019, 8461725. [Google Scholar] [CrossRef]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Sarawi, W.; Attia, S.M.; Alanazi, W.A.; Ibrahim, K.E.; Alsanea, S.; Alqarni, S.A. Acetyl-11-keto-β-boswellic acid improves clinical symptoms through modulation of Nrf2 and NF-κB pathways in SJL/J mouse model of experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2022, 107, 108703. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Lowell, J.A. Th17 cells in depression. Brain Behav. Immun. 2018, 69, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Zelinski, E.L.; Schubert, C.L.; Knight, D.; Craig, L.A.; Winston, B.W.; Spanswick, S.C.; Petri, B.; Jenne, C.N.; Sutherland, J.C.; et al. Targeting inflammatory monocytes in sep-sis-associated encephalopathy and long-term cognitive impairment. JCI Insight. 2018, 3, 99364. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Peng, S.; Lin, K.; Bin Zhao, B.; Wei, L.; Tuo, Q.; Liao, D.; Yuan, T.; Shi, Z. Chronic stress-induced depression requires the recruitment of peripheral Th17 cells into the brain. J. Neuroinflammation 2022, 19, 186. [Google Scholar] [CrossRef]
- Alhazzani, K.; Ahmad, S.F.; Al-Harbi, N.O.; Attia, S.M.; Bakheet, S.A.; Sarawi, W.; Alqarni, S.A.; Algahtani, M.; Nadeem, A. Pharmacological Inhibition of STAT3 by Stattic Ameliorates Clinical Symptoms and Reduces Autoinflammation in Myeloid, Lymphoid, and Neuronal Tissue Compartments in Relapsing-Remitting Model of Experimental Autoimmune Encephalomyelitis in SJL/J Mice. Pharmaceutics 2021, 13, 925. [Google Scholar] [CrossRef]
- Gomez-Rodriguez, J.; Wohlfert, E.A.; Handon, R.; Meylan, F.; Wu, J.Z.; Anderson, S.M.; Kirby, M.R.; Belkaid, Y.; Schwartzberg, P.L. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J. Exp. Med. 2014, 211, 529–543. [Google Scholar] [CrossRef]
- Jiang, Q.; Yang, G.; Liu, Q.; Wang, S.; Cui, D. Function and Role of Regulatory T Cells in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 626193. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Li, Y.; Feng, Y.-F.; Liu, X.-T.; Li, Y.-C.; Zhu, H.-M.; Sun, M.-R.; Li, P.; Liu, B.; Yang, H. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 2021, 38, 101771. [Google Scholar] [CrossRef]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Bio-Chem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Thimmulappa, R.; Craciun, F.; Harvey, C.; Singh, A.; Kombairaju, P.; Reddy, S.P.; Remick, D.; Biswal, S. Enhancing Nrf2 Pathway by Disruption of Keap1 in Myeloid Leukocytes Protects against Sepsis. Am. J. Respir. Crit. Care Med. 2011, 184, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, S.G.; Bailey, M.T.; Sheridan, J.F.; Padgett, D.A.; Avitsur, R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav. Immun. 2007, 21, 458–466. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Nadeem, A.; Ansari, M.A.; Al-Harbi, M.M.; Alotaibi, M.R.; AlSaad, A.M.; Ahmad, S.F. Psoriasis-like inflammation leads to renal dysfunction via upregulation of NADPH oxidases and inducible nitric oxide synthase. Int. Immunopharmacol. 2017, 46, 1–8. [Google Scholar] [CrossRef]
- Nadeem, A.; Al-Harbi, N.O.; Al-Harbi, M.M.; El-Sherbeeny, A.M.; Ahmad, S.F.; Siddiqui, N.; Ansari, M.A.; Zoheir, K.M.; Attia, S.M.; Al-Hosaini, K.A.; et al. Imiquimod-induced psoriasis-like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL-17A pathway modulation. Pharmacol. Res. 2015, 99, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Chhabra, S.K.; Masood, A.; Raj, H.G. Increased oxidative stress and altered levels of antioxidants in asthma. J. Allergy Clin. Immunol. 2003, 111, 72–78. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Algahtani, M.M.; Alshehri, S.; Alqarni, S.S.; Ahmad, S.F.; Al-Harbi, N.O.; Alqarni, S.A.; Alfardan, A.S.; Ibrahim, K.E.; Attia, S.M.; Nadeem, A. Inhibition of ITK Signaling Causes Amelioration in Sepsis-Associated Neuroinflammation and Depression-like State in Mice. Int. J. Mol. Sci. 2023, 24, 8101. https://doi.org/10.3390/ijms24098101
Algahtani MM, Alshehri S, Alqarni SS, Ahmad SF, Al-Harbi NO, Alqarni SA, Alfardan AS, Ibrahim KE, Attia SM, Nadeem A. Inhibition of ITK Signaling Causes Amelioration in Sepsis-Associated Neuroinflammation and Depression-like State in Mice. International Journal of Molecular Sciences. 2023; 24(9):8101. https://doi.org/10.3390/ijms24098101
Chicago/Turabian StyleAlgahtani, Mohammad M., Samiyah Alshehri, Sana S. Alqarni, Sheikh F. Ahmad, Naif O. Al-Harbi, Saleh A. Alqarni, Ali S. Alfardan, Khalid E. Ibrahim, Sabry M. Attia, and Ahmed Nadeem. 2023. "Inhibition of ITK Signaling Causes Amelioration in Sepsis-Associated Neuroinflammation and Depression-like State in Mice" International Journal of Molecular Sciences 24, no. 9: 8101. https://doi.org/10.3390/ijms24098101
APA StyleAlgahtani, M. M., Alshehri, S., Alqarni, S. S., Ahmad, S. F., Al-Harbi, N. O., Alqarni, S. A., Alfardan, A. S., Ibrahim, K. E., Attia, S. M., & Nadeem, A. (2023). Inhibition of ITK Signaling Causes Amelioration in Sepsis-Associated Neuroinflammation and Depression-like State in Mice. International Journal of Molecular Sciences, 24(9), 8101. https://doi.org/10.3390/ijms24098101