
Combined BCL-2 and PI3K/AKT Pathway Inhibition in KMT2A-Rearranged Acute B-Lymphoblastic Leukemia Cells
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Co-Targeting BCL-2 and PI3K/AKT Pathways Synergistically Reduces Blast Viability While Sparing Healthy Blood Cells
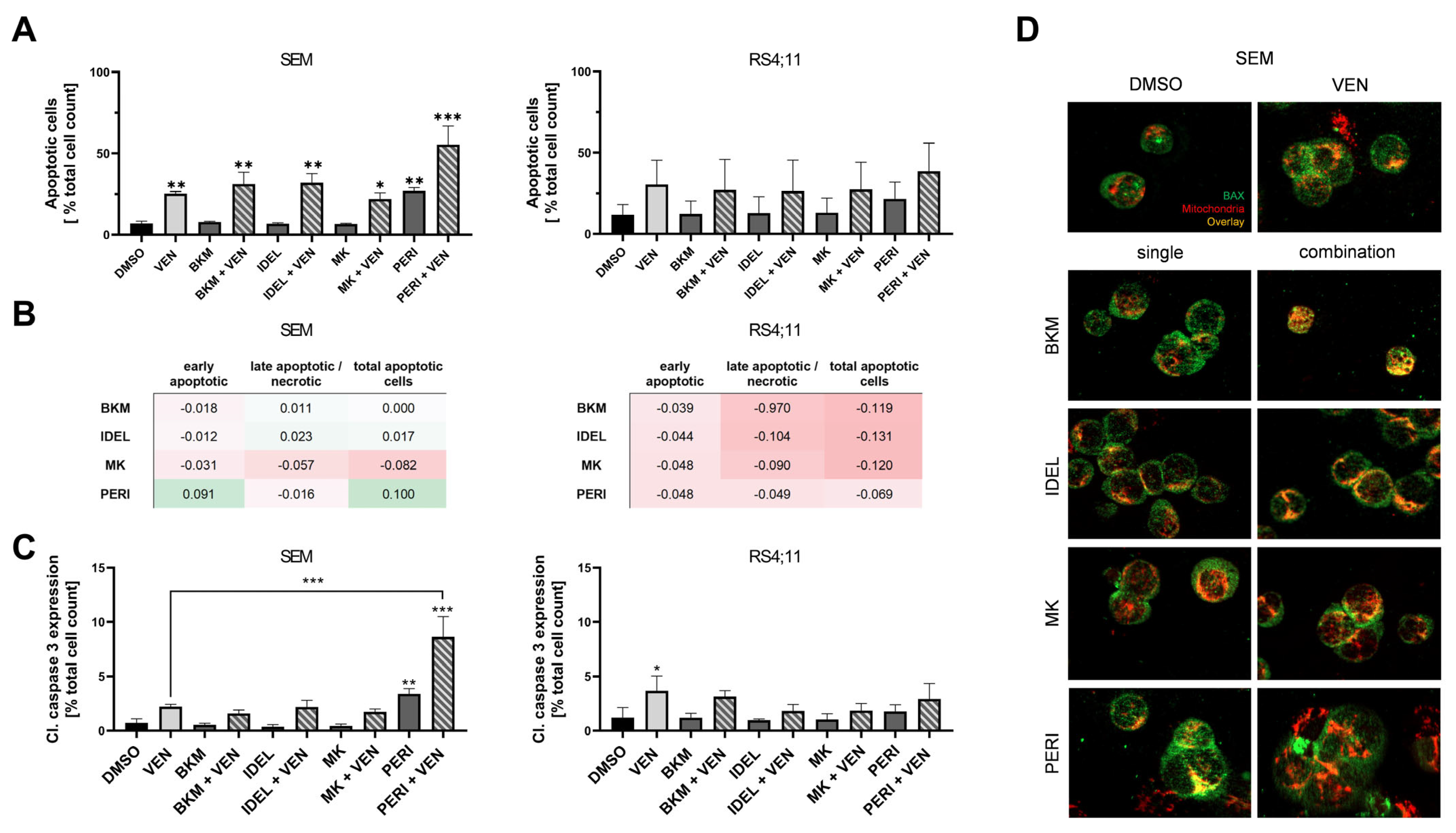
2.2. Combined Application of VEN and PERI Potentiates Caspase-3 Activation and Synergistically Induces Apoptosis
2.3. AKT Phosphorylation Correlates with Anti-Apoptotic Protein Expression and Activity
2.4. PERI Results in a Pro-Apoptotic Modulation of the BCL-2 Pathway That Includes Significant BBC3 Enhancement
2.5. PI3K/AKT Inhibitors Cause Modulation of the Extrinsic Apoptosis Signaling Cascade
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Inhibitors
4.2. Inhibitory Experiments
4.3. Morphology Analyses
4.4. Analysis of Apoptotic Processes
4.5. Intracellular Flow-Cytometric Protein Expression Measurement
4.6. Targeted RNA Custom Panel Sequencing
4.7. Immunoblot
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malard, F.; Mohty, M. Acute Lymphoblastic Leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Khaw, S.L.; Suryani, S.; Evans, K.; Richmond, J.; Robbins, A.; Kurmasheva, R.T.; Billups, C.A.; Erickson, S.W.; Guo, Y.; Houghton, P.J.; et al. Venetoclax Responses of Pediatric ALL Xenografts Reveal Sensitivity of MLL-Rearranged Leukemia. Blood 2016, 128, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, A.; Lange, S.; Holz, C.; Brock, L.; Freitag, T.; Sekora, A.; Knuebel, G.; Krohn, S.; Schwarz, R.; Hinz, B.; et al. Effective Tumor Cell Abrogation via Venetoclax-Mediated BCL-2 Inhibition in KMT2A-Rearranged Acute B-Lymphoblastic Leukemia. Cell Death Discov. 2022, 8, 302. [Google Scholar] [CrossRef]
- Benito, J.M.; Godfrey, L.; Kojima, K.; Hogdal, L.; Wunderlich, M.; Geng, H.; Marzo, I.; Harutyunyan, K.G.; Golfman, L.; North, P.; et al. MLL-Rearranged Acute Lymphoblastic Leukemias Activate BCL-2 through H3K79 Methylation and Are Sensitive to the BCL-2-Specific Antagonist ABT-199. Cell Rep. 2015, 13, 2715–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From Basic Apoptosis Discoveries to Advanced Selective BCL-2 Family Inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Valentin, R.; Grabow, S.; Davids, M.S. The Rise of Apoptosis: Targeting Apoptosis in Hematologic Malignancies. Blood 2018, 132, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Debatin, K.M. Extrinsic versus Intrinsic Apoptosis Pathways in Anticancer Chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. NIH: U.S. National Library of Medicine, ClinicalTrials.Gov, Venetoclax in ALL. Available online: https://Clinicaltrials.Gov/Ct2/Results?Cond=Acute+Lymphoblastic+Leukemia&term=Venetoclax&cntry=&state=&city=&dist= (accessed on 7 September 2022).
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef]
- Kapoor, I.; Bodo, J.; Hill, B.T.; Hsi, E.D.; Almasan, A. Targeting BCL-2 in B-Cell Malignancies and Overcoming Therapeutic Resistance. Cell Death Dis. 2020, 11, 941. [Google Scholar] [CrossRef]
- Fidyt, K.; Pastorczak, A.; Cyran, J.; Crump, N.T.; Goral, A.; Madzio, J.; Muchowicz, A.; Poprzeczko, M.; Domka, K.; Komorowski, L.; et al. Potent, P53-Independent Induction of NOXA Sensitizes MLL-Rearranged B-Cell Acute Lymphoblastic Leukemia Cells to Venetoclax. Oncogene 2022, 41, 1600–1609. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Neri, L.M.; Cani, A.; Martelli, A.M.; Simioni, C.; Junghanss, C.; Tabellini, G.; Ricci, F.; Tazzari, P.L.; Pagliaro, P.; Mccubrey, J.A. Targeting the PI3K/Akt/MTOR Signaling Pathway in B-Precursor Acute Lymphoblastic Leukemia and Its Therapeutic Potential. Leukemia 2014, 28, 739–748. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A Phosphatidylinositol 3-Kinase/Akt Pathway Promotes Translocation of Mdm2 from the Cytoplasm to the Nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.R.; Dudek, H.; Xu, T.; Masters, S.; Haian, F.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples Survival Signals to the Cell- Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen Synthase Kinase-3 Regulates Mitochondrial Outer Membrane Permeabilization and Apoptosis by Destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Vitale, N.; Coma, S.; Arruga, F.; Chadburn, A.; di Napoli, A.; Laudanna, C.; Allan, J.N.; Furman, R.R.; Pachter, J.A.; et al. Synergistic Efficacy of the Dual PI3K-δ/γ Inhibitor Duvelisib with the Bcl-2 Inhibitor Venetoclax in Richter Syndrome PDX Models. Blood 2021, 137, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [Green Version]
- Bojarczuk, K.; Wienand, K.; Ryan, J.A.; Chen, L.; Villalobos-Ortiz, M.; Mandato, E.; Stachura, J.; Letai, A.; Lawton, L.N.; Chapuy, B.; et al. Targeted Inhibition of PI3Ka/d Is Synergistic with BCL-2 Blockade in Genetically Defined Subtypes of DLBCL. Blood 2019, 133, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Crassini, K.R.; Fatima, N.; Christopherson, R.; Mulligan, S.P.; Best, O.G. Venetoclax Is Synergistic with Idelalisib or MK2206 Against Primary CLL Cells in an in Vitro Model of the Microenvironment. Blood 2019, 134, 5443. [Google Scholar] [CrossRef]
- Haselager, M.V.; Kielbassa, K.; ter Burg, J.; Bax, D.J.C.; Fernandes, S.M.; Borst, J.; Tam, C.; Forconi, F.; Chiodin, G.; Brown, J.R.; et al. Changes in Bcl-2 Members after Ibrutinib or Venetoclax Uncover Functional Hierarchy in Determining Resistance to Venetoclax in CLL. Blood 2020, 136, 2918–2926. [Google Scholar] [CrossRef]
- Friedman, D.R.; Lanasa, M.C.; Davis, P.H.; Allgood, S.D.; Matta, K.M.; Brander, D.M.; Chen, Y.; Davis, E.D.; Volkheimer, A.D.; Moore, J.O.; et al. Perifosine Treatment in Chronic Lymphocytic Leukemia: Results of a Phase II Clinical Trial and in Vitro Studies. Leuk. Lymphoma 2014, 55, 1067–1075. [Google Scholar] [CrossRef]
- Oki, Y.; Fanale, M.; Romaguera, J.; Fayad, L.; Fowler, N.; Copeland, A.; Samaniego, F.; Kwak, L.W.; Neelapu, S.; Wang, M.; et al. Phase II Study of an AKT Inhibitor MK2206 in Patients with Relapsed or Refractory Lymphoma. Br. J. Haematol. 2015, 171, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Assouline, S.; Amrein, L.; Aloyz, R.; Banerji, V.; Caplan, S.; Owen, C.; Hasegawa, W.; Robinson, S.; Shivakumar, S.; Prica, A.; et al. IND.216: A Phase II Study of Buparlisib and Associated Biomarkers, Raptor and P70S6K, in Patients with Relapsed and Refractory Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2020, 61, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.; Loscocco, F.; Visani, G.; Paolasini, S.; Scalzulli, P.; Musto, P.; Perrone, T.; Guarini, A.; Pastore, D.; Mazza, P.; et al. Real-Life Efficacy and Safety of Idelalisib in 55 Double-Refractory Follicular Lymphoma Patients. Br. J. Haematol. 2022, 199, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-XL-Dependent Resistance to the BCL-2 Inhibitor ABT-199 Can Be Overcome by Preventing PI3K/AKT/MTOR Activation in Lymphoid Malignancies. Cell Death Dis. 2015, 6, e1593-12. [Google Scholar] [CrossRef] [Green Version]
- Naderali, E.; Valipour, B.; Khaki, A.A.; Soleymani Rad, J.; Alihemmati, A.; Rahmati, M.; Nozad Charoudeh, H. Positive Effects of PI3K/Akt Signaling Inhibition on PTEN and P53 in Prevention of Acute Lymphoblastic Leukemia Tumor Cells. Adv. Pharm. Bull. 2019, 9, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Safaroghli-azar, A.; Bashash, D.; Sadreazami, P.; Momeny, M.; Ghaffari, S.H. PI3K- δ Inhibition Using CAL-101 Exerts Apoptotic Effects and Increases Doxorubicin-Induced Cell Death in Pre-B-Acute Lymphoblastic Leukemia Cells. Anticancer Drugs 2017, 28, 436–445. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Walter, R.B.; Faderl, S.H.; Jabbour, E.J.; Zeng, Z.; Borthakur, G.; Huang, X.; Kadia, T.M.; Ruvolo, P.P.; Feliu, J.B.; et al. Preclinical and Early Clinical Evaluation of the Oral AKT Inhibitor, MK-2206, for the Treatment of Acute Myelogenous Leukemia. Clin. Cancer Res. 2014, 20, 2226–2235. [Google Scholar] [CrossRef]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a Novel Akt Inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [Google Scholar] [CrossRef]
- Richter, A.; Fischer, E.; Holz, C.; Schulze, J.; Lange, S.; Sekora, A.; Knuebel, G.; Henze, L.; Roolf, C.; Escobar, H.M.; et al. Combined Application of Pan-Akt Inhibitor Mk-2206 and Bcl-2 Antagonist Venetoclax in b-Cell Precursor Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2771. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt Inhibitors in Cancer Treatment: The Long Journey from Drug Discovery to Clinical Use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragon, B.; Kantarjian, H.; Jabbour, E.; Ravandi, F.; Cortes, J.; Borthakur, G.; DeBose, L.; Zeng, Z.; Schneider, H.; Pemmaraju, N.; et al. Buparlisib, a PI3K Inhibitor, Demonstrates Acceptable Tolerability and Preliminary Activity in a Phase I Trial of Patients with Advanced Leukemiass. Am. J. Hematol. 2017, 92, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Catley, L.; Yasui, H.; Ishitsuka, K.; Raje, N.; Mitsiades, C.; Podar, K.; Munshi, N.C.; Chauhan, D.; Richardson, P.G.; et al. Perifosine, an Oral Bioactive Novel Alkylphospholipid, Inhibits Akt and Induces in Vitro and in Vivo Cytotoxicity in Human Multiple Myeloma Cells. Blood 2006, 107, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarini, F.; del Sole, M.; Mongiorgi, S.; Gaboardi, G.C.; Cappellini, A.; Mantovani, I.; Follo, M.Y.; Mccubrey, J.A.; Martelli, A.M. The Novel Akt Inhibitor, Perifosine, Induces Caspase-Dependent Apoptosis and Downregulates P-Glycoprotein Expression in Multidrug-Resistant Human T-Acute Leukemia Cells by a JNK-Dependent Mechanism. Leukemia 2008, 22, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Papa, V.; Tazzari, P.L.; Chiarini, F.; Cappellini, A.; Ricci, F.; Billi, A.M.; Evangelisti, C.; Ottaviani, E.; Giovanni, M.; Testoni, N.; et al. Proapoptotic Activity and Chemosensitizing Effect of the Novel Akt Inhibitor Perifosine in Acute Myelogenous Leukemia Cells. Leukemia 2008, 22, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Xu, L.; Zhao, Q. Perifosine and ABT-737 Synergistically Inhibit Lung Cancer Cells in Vitro and in Vivo. Biochem. Biophys. Res. Commun. 2016, 473, 1170–1176. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Green, D.R. PUMA Cooperates with Direct Activator Proteins to Promote Mitochondrial Outer Membrane Permeabilization and Apoptosis. Cell Cycle 2009, 8, 2692–2696. [Google Scholar] [CrossRef] [Green Version]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. P53- and Drug-Induced Apoptotic Responses Mediated by BH3-Only Proteins Puma and Noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef]
- Yu, H.J.; Ahn, C.H.; Yang, I.H.; Won, D.H.; Jin, B.; Cho, N.P.; Hong, S.D.; Shin, J.A.; Cho, S.D. Apoptosis Induced by Methanol Extract of Potentilla Discolor in Human Mucoepidermoid Carcinoma Cells through STAT3/PUMA Signaling Axis. Mol. Med. Rep. 2018, 17, 5258–5264. [Google Scholar] [CrossRef]
- Amente, S.; Zhang, J.; Lubrano Lavadera, M.; Lania, L.; Avvedimento, E.V.; Majello, B. Myc and PI3K/AKT Signaling Cooperatively Repress FOXO3a-Dependent PUMA and GADD45a Gene Expression. Nucleic Acids Res. 2011, 39, 9498–9507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.Q.; Shen, M.; Wu, W.J.; Li, B.J.; Weng, Q.N.; Li, M.; Liu, H.L. Expression of PUMA in Follicular Granulosa Cells Regulated by FoxO1 Activation During Oxidative Stress. Reprod. Sci. 2015, 22, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ji, S.-Y.; Yang, J.-L.; Li, X.-X.; Zhang, J.; Zhang, Y.; Hu, Z.-Y.; Liu, Y.-X. Wnt/β-Catenin Signaling Regulates Follicular Development by Modulating the Expression of Foxo3a Signaling Components. Mol. Cell Endocrinol. 2014, 382, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Akhter, R.; Sanphui, P.; Biswas, S.C. The Essential Role of P53-up-Regulated Modulator of Apoptosis (Puma) and Its Regulation by FoxO3a Transcription Factor in β-Amyloid-Induced Neuron Death. J. Biol. Chem. 2014, 289, 10812–10822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 Determines the Induction of PUMA and Apoptosis by P53. Mol. Cell 2011, 42, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Schubert, F.; Rapp, J.; Brauns-Schubert, P.; Schlicher, L.; Stock, K.; Wissler, M.; Weiß, M.; Charvet, C.; Borner, C.; Maurer, U. Requirement of GSK-3 for PUMA Induction upon Loss of pro-Survival PI3K Signaling Article. Cell Death Dis. 2018, 9, 470. [Google Scholar] [CrossRef] [Green Version]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A Elicits Apoptosis through an MST2 Pathway Directing Proapoptotic Transcription by the P73 Tumor Suppressor Protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-X.; Ma, D.-Y.; Yao, Z.-Q.; Fu, C.-Y.; Shi, Y.-X.; Wang, Q.-L. ERK1/2/P53 and NF-KB Dependent-PUMA Activation Involves in Doxorubicin-Induced Cardiomyocyte Apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2435–2442. [Google Scholar]
- Akhter, R.; Sanphui, P.; Das, H.; Saha, P.; Biswas, S.C. The Regulation of P53 Up-Regulated Modulator of Apoptosis by JNK/c-Jun Pathway in β-Amyloid-Induced Neuron Death. J. Neurochem. 2015, 134, 1091–1103. [Google Scholar] [CrossRef]
- Gomez-Lazaro, M.; Galindo, M.F.; Concannon, C.G.; Segura, M.F.; Fernandez-Gomez, F.J.; Llecha, N.; Comella, J.X.; Prehn, J.H.M.; Jordan, J. 6-Hydroxydopamine Activates the Mitochondrial Apoptosis Pathway through P38 MAPK-Mediated, P53-Independent Activation of Bax and PUMA. J. Neurochem. 2008, 104, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jung, Y.S.; Chung, J.Y.; Oh, A.Y.; Lee, S.J.; Choi, D.H.; Jang, S.M.; Jang, K.S.; Paik, S.S.; Ha, N.C.; et al. Novel Tumor Suppressive Function of Smad4 in Serum Starvation-Induced Cell Death through PAK1-PUMA Pathway. Cell Death Dis. 2011, 2, e235. [Google Scholar] [CrossRef] [Green Version]
- Spender, L.C.; Carter, M.J.; O’Brien, D.I.; Clark, L.J.; Yu, J.; Michalak, E.M.; Happo, L.; Cragg, M.S.; Inman, G.J. Transforming Growth Factor-β Directly Induces P53-up-Regulated Modulator of Apoptosis (PUMA) during the Rapid Induction of Apoptosis in Myc-Driven B-Cell Lymphomas0. J. Biol. Chem. 2013, 288, 5198–5209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, I.M.; Roccaro, A.; Hong, F.; Weller, E.; Rubin, N.; Leduc, R.; Rourke, M.; Chuma, S.; Sacco, A.; Jia, X.; et al. Clinical and Translational Studies of a Phase II Trial of the Novel Oral Akt Inhibitor Perifosine in Relapsed or Relapsed/Refractory Waldenström’s Macroglobulinemia. Clin. Cancer Res. 2010, 16, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holz, C.; Lange, S.; Sekora, A.; Knuebel, G.; Krohn, S.; Murua Escobar, H.; Junghanss, C.; Richter, A. Combined BCL-2 and PI3K/AKT Pathway Inhibition in KMT2A-Rearranged Acute B-Lymphoblastic Leukemia Cells. Int. J. Mol. Sci. 2023, 24, 1359. https://doi.org/10.3390/ijms24021359
Holz C, Lange S, Sekora A, Knuebel G, Krohn S, Murua Escobar H, Junghanss C, Richter A. Combined BCL-2 and PI3K/AKT Pathway Inhibition in KMT2A-Rearranged Acute B-Lymphoblastic Leukemia Cells. International Journal of Molecular Sciences. 2023; 24(2):1359. https://doi.org/10.3390/ijms24021359
Chicago/Turabian StyleHolz, Clemens, Sandra Lange, Anett Sekora, Gudrun Knuebel, Saskia Krohn, Hugo Murua Escobar, Christian Junghanss, and Anna Richter. 2023. "Combined BCL-2 and PI3K/AKT Pathway Inhibition in KMT2A-Rearranged Acute B-Lymphoblastic Leukemia Cells" International Journal of Molecular Sciences 24, no. 2: 1359. https://doi.org/10.3390/ijms24021359
APA StyleHolz, C., Lange, S., Sekora, A., Knuebel, G., Krohn, S., Murua Escobar, H., Junghanss, C., & Richter, A. (2023). Combined BCL-2 and PI3K/AKT Pathway Inhibition in KMT2A-Rearranged Acute B-Lymphoblastic Leukemia Cells. International Journal of Molecular Sciences, 24(2), 1359. https://doi.org/10.3390/ijms24021359