
Redox-Cycling “Mitocans” as Effective New Developments in Anticancer Therapy

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
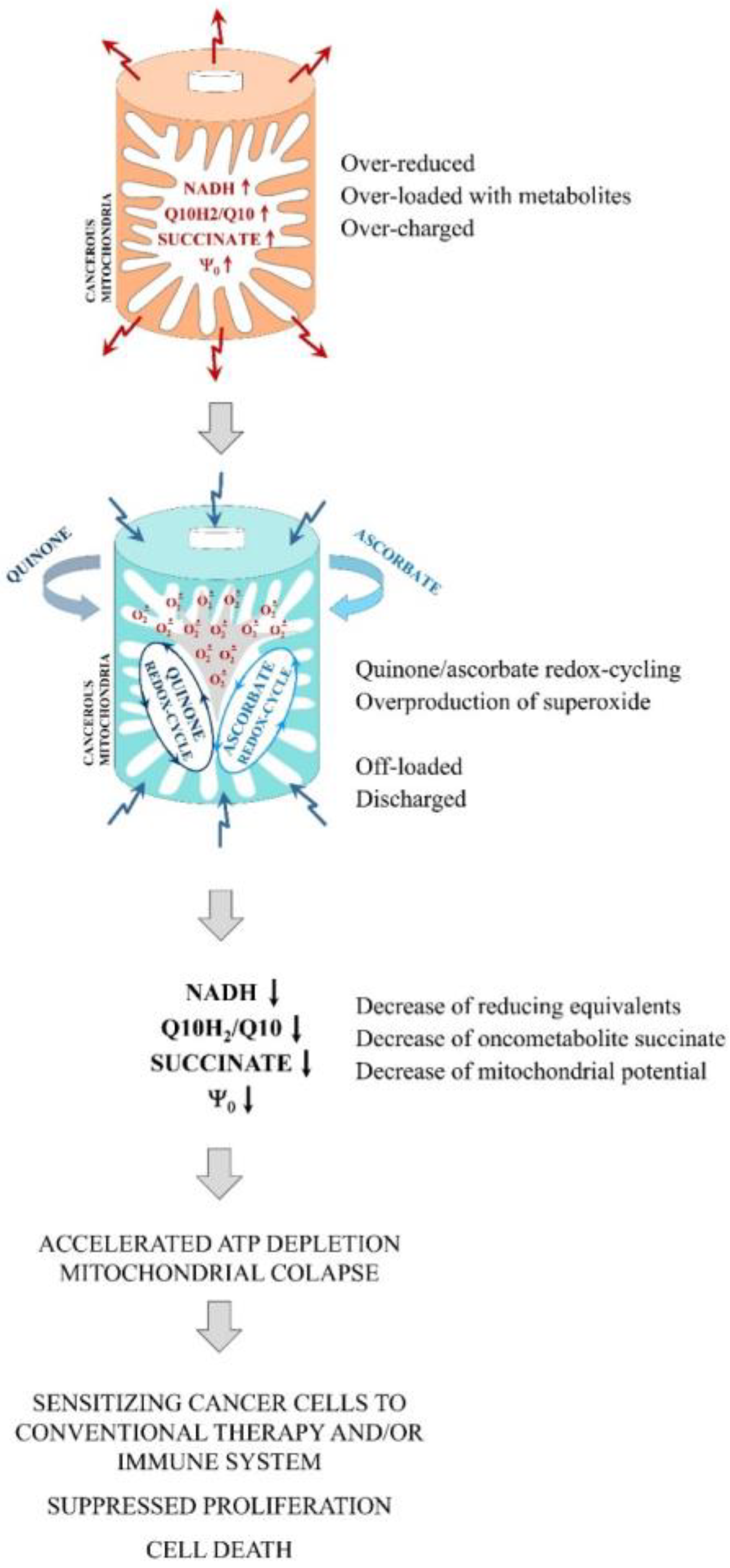
:1. Introduction
2. Results
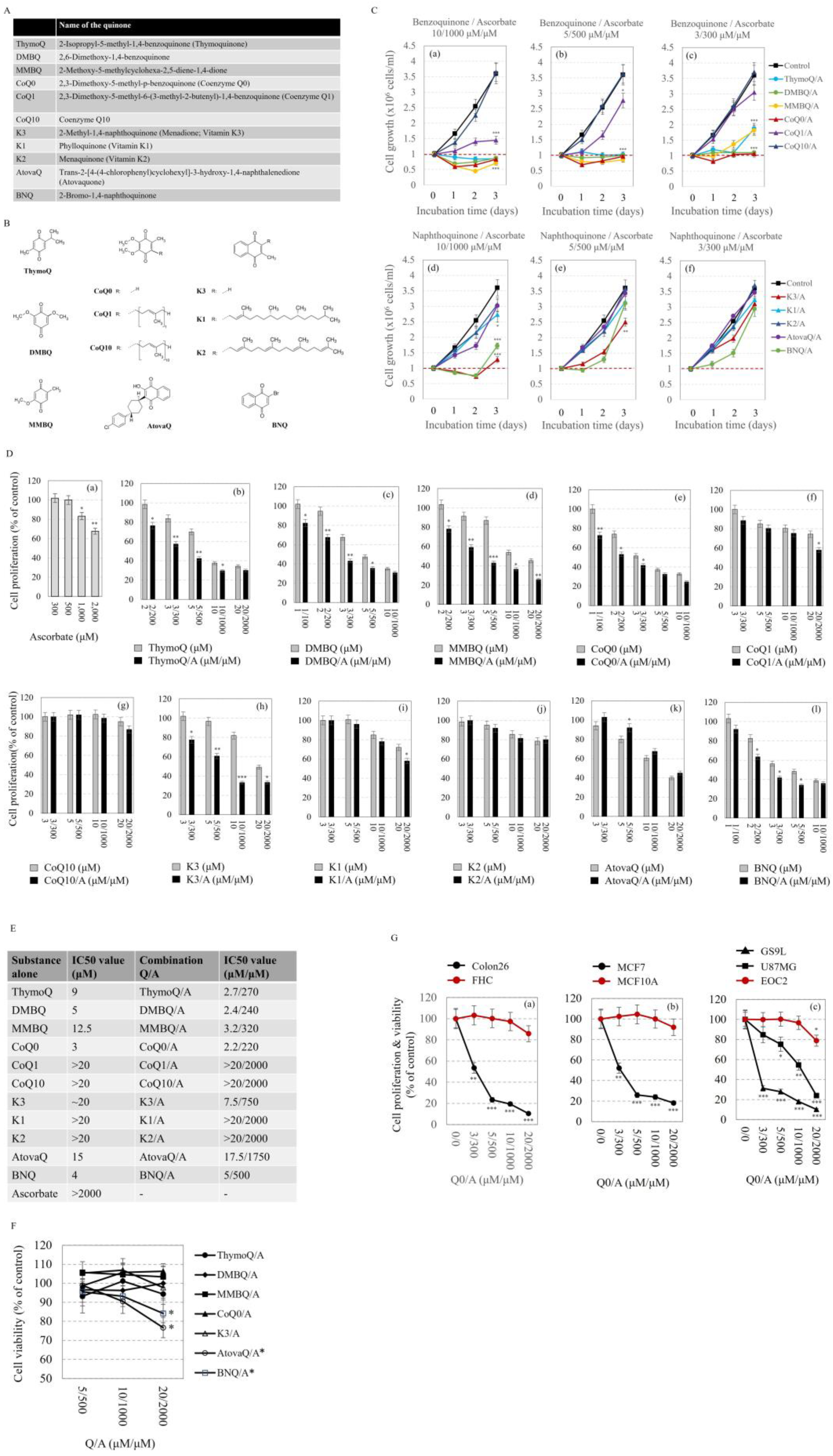
2.1. Effects of Q/A Redox-Pairs on Cell Proliferation and Viability In Vitro
- (1)
- (2)
- The Q/A redox-pairs containing unprenylated benzoquinones exhibited marked cytotoxicity within 48 h of incubation (Figure 2C(c)). In this case, the number of live cells in the Q/A-treated samples dropped below the initial level at: 3/300 μM/μM for CoQ0/A; 5/500 μM/μM for CoQ0/A and MMBQ/A; and 10/1000 μM/μM for all unprenylated benzoquinones. In the naphthoquinone group, K3/A and BNQ/A also exhibited cytotoxicity, but only at 10/1000 μM/μM (Figure 2C(d)).
- (3)
- The antiproliferative effect of the Q/A redox-pairs containing prenylated quinones (CoQ10, K1, K2) was negligible, while that of containing unprenylated quinones (CoQ0, K3, DMBQ, MMBQ, BNQ, ThymoQ) was markedly expressed even at low concentrations of Q/A such as 2/200 μM/μM and 3/300 μM/μM. In the coenzyme Q group, the shortening of the isoprenoid chain increased the antiproliferative activity in the order: CoQ0/A > CoQ1/A > CoQ10/A, which was very well seen at 10/1000 and 20/2000 μM/μM (Figure 2C(a,b),D).
- (4)
- Antiproliferative and cytotoxic effects were most pronounced for the CoQ0/A and DMBQ/A.
2.2. Effects of Q/A Redox-Pairs on Mitochondrial Oxidative Stress and Steady-State ATP In Vitro
2.3. Effects of Q/A Redox-Pair on Tumor Growth and Tissue Redox-State In Vivo
2.4. Summary of the Main Experimental Findings
- (1)
- Q/A redox-pairs containing unprenylated quinones were characterized by pronounced antiproliferative and cytotoxic effects on cancer cells. This was accompanied by a dose-dependent overproduction of mitochondrial superoxide and accelerated ATP depletion. Q/A redox-pairs containing prenylated quinones, especially long-chain ones, did not significantly affect cancer cell proliferation and viability, as well as mitochondrial oxidative stress and energy balance. Their effects on these parameters were negligible.
- (2)
- Q/A redox-pairs containing quinones such as CoQ0, DMBQ, MMBQ, and K3 did not significantly affect the viability of normal cells and steady-state level of ATP in them. These redox-pairs induced mild and dose-independent mitochondrial oxidative stress in normal cells, which seems to be well tolerated.
- (3)
- The anticancer effects of the investigated benzoquinone/ascorbate redox-pairs were more pronounced than those of naphthoquinone/ascorbate redox-pairs. The most effective was CoQ0/A followed by DMBQ/A, as both redox-pairs are a better alternative to the widely investigated K3/A.
- (4)
- CoQ0/A was more effective than K3/A in suppressing tumor growth and increasing survival in the investigated mouse models (glioblastoma and colon cancer xenografts). CoQ0/A decreased the size of the tumor, while K3/A caused only a growth arrest (p < 0.05). Both redox-pairs did not induce adverse drug-related side-effects such as fever, tetraplegia, convulsions, etc., that are characteristic of many conventional anticancer drugs.
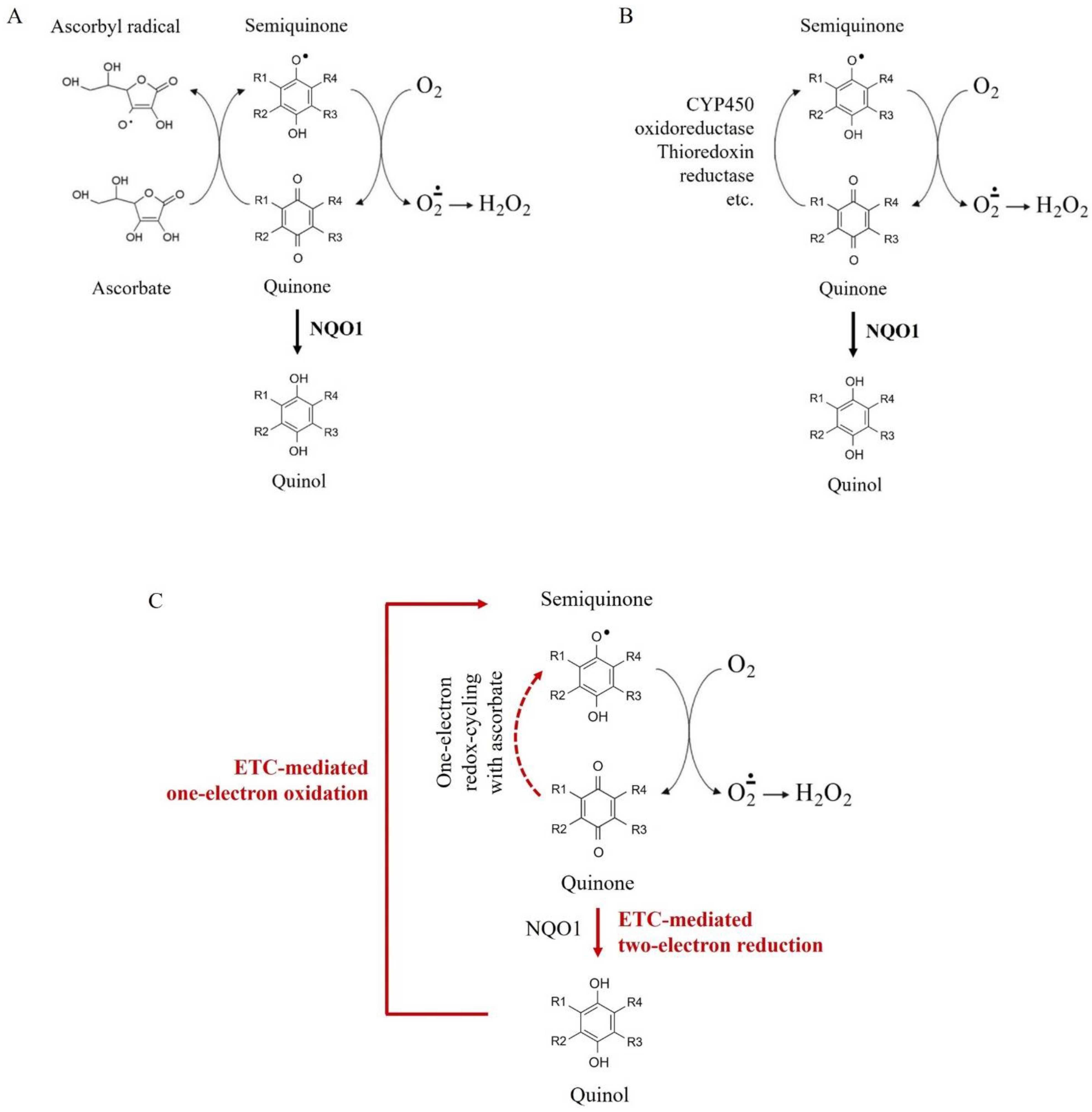
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cells and Treatment Protocol
4.3. Cell Viability and Proliferation Assay
4.4. Mitochondrial Superoxide Assay
4.5. ATP Assay
4.6. Total Antioxidant Capacity (TAC) Assay
4.7. tNOX (ENOX2) Assay
4.8. Animals and Treatment Protocols
4.9. In Vivo MRI Measurements
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mkhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced response in breast cancers. Semin. Cancer Biol. 2022, 83, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Ralf, S.J.; Low, P.; Dong, L.; Lawen, A.; Neuzil, J. Mitocans: Mitochondrial targeted anti-cancer drugs as improved therapies and related patient documents. Recent Pat. Anticancer Drug. Discov. 2006, 1, 327–346. [Google Scholar]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans revisited: Mitochondrial targeting as efficient anti-cancer therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef]
- Verrax, J.; Beck, R.; Dejeans, R.B.; Glorieux, C.; Sid, B.; Curi Pedrosa, R.; Benites, J.; Vasquez, D.; Valderrama, J.A.; Buc Calderon, P. Redox-active quinones and ascorbate: An innovative cancer therapy that exploits the vulnerability of cancer cells to oxidative stress. Anticancer Agents Med. Chem. 2011, 11, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Otto, A.; Hahlbrock, T.; Eich, K.; Karaaslan, F.; Jurgens, C.; Germer, C.-T.; Wiegering, A.; Kammerer, U. Antiproliferative and antimetabolic effects behind the anticancer property of fermented wheat germ extract. BMC Complement. Altern. Med. 2016, 16, 160. [Google Scholar] [CrossRef]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva-Jr, F.P. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell. Int. 2019, 19, 207. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Santhosh, S.M.; Coppo, L.; Ogata, F.T.; Lu, J.; Holmgren, A. The combination of ascorbate and menadione causes cancer cell death by oxidative stress and replicative stress. Free Radic. Biol. Med. 2019, 134, 350–358. [Google Scholar] [CrossRef]
- Bajor, M.; Graczyk-Jarzynka, A.; Marhelava, K.; Kurkowiak, M.; Rahman, A.; Aura, C.; Russell, N.; Zych, A.O.; Firczuk, M.; Winiarska, M.; et al. Triple combination of ascorbate, menadione and inhibition of peroxiredoxin-1 produces synergistic cytotoxic effects in triple-negative breast cancer cells. Antioxidants 2020, 9, 320. [Google Scholar] [CrossRef]
- Bakalova, R.; Semkova, S.; Ivanova, D.; Zhelev, Z.; Miller, T.; Takeshima, T.; Shibata, S.; Lazarova, D.; Aoki, I.; Higashi, T. Selective targeting of cancerous mitochondria and suppression of tumor growth using redox-active treatment adjuvant. Oxid. Med. Cell. Longev. 2020, 2020, 6212935. [Google Scholar] [CrossRef]
- Kapur, A.; Mehta, P.; Simmons, A.D.; Eriksen, S.S.; Mehta, G.; Palecek, S.P.; Felder, M.; Stenerson, Z.; Nayak, A.; Dominguez, J.M.A.; et al. Atovaquone: An inhibitor of oxidative phosphorylation as studied in gynecologic cancers. Cancers 2022, 14, 2297. [Google Scholar] [CrossRef]
- Sumiyoshi, A.; Shibata, S.; Zhelev, Z.; Miller, T.; Lazarova, D.; Aoki, I.; Obata, T.; Higashi, T.; Bakalova, R. Targeting glioblastoma via selective alteration of mitochondrial redox-state. Cancers 2022, 14, 485. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Teng, S.; Wilson, J.X.; Galati, G.; Khan, S.; O’Brien, P.J. Coenzyme Q cytoprotective metabolism for mitochondrial complex I cytopathies involves NAD(P)H:quinone oxidoreductase (NQO1). Free Radic. Res. 2002, 36, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Sarewicz, M.; Osyczka, A. Electronic connection between the quinone and cytochrome c redox pools and its role in regulation of mitochondrial electron transport and redox signaling. Physiol. Rev. 2015, 95, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mevers, E.; Higgins, K.W.; Fomina, Y.; Zhang, J.; Mandinova, A.; Newman, D.; Shaw, S.Y.; Clardy, J.; Mootha, V.K. Natural product screening reveals naphthoquinone complex I bypass factors. PLoS ONE 2016, 11, e0162686. [Google Scholar] [CrossRef]
- Collins, R.R.J.; Patel, K.; Putnam, W.C.; Kapur, P.; Rakheja, D. Oncometabolites: A new paradigm for oncology, metabolism, the clinical laboratory. Clin. Chem. 2017, 63, 1812–1820. [Google Scholar] [CrossRef]
- Tielens, A.G.M.; Rotte, C.; van Hellemond, J.J.; Martin, W. Mitochondria as we don’t know them. Trends Biochem. Sci. 2002, 27, 564–572. [Google Scholar] [CrossRef]
- Dobouchaud, H.; Walter, L.; Rigoulet, M.; Batandier, C. Mitochondrial NADH redox potential impacts the reactive oxygen species production of reverse electron transfer through complex I. J. Bioenerg. Biomembr. 2018, 50, 367–377. [Google Scholar] [CrossRef]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, T.A.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef]
- Bonnilla-Porras, A.R.; Jimenez-Del-Rio, M.; Valez-Pardo, C. Vitamin K3 and vitamin C alone or in combination induced apoptosis in leukemia cells by similar oxidative stress signaling mechanism. Cancer Cell. Int. 2011, 11, 19. [Google Scholar] [CrossRef]
- Verrax, J.; Pedrosa, R.C.; Beck, R.; Dejeans, N.; Taper, H.; Calderon, P.B. In situ modulation of oxidative stress: A novel and efficient strategy to kill cancer cells. Curr. Med. Chem. 2009, 6, 182130. [Google Scholar] [CrossRef]
- Tareen, B.; Summers, J.L.; Jamison, J.M.; Neal, D.R.; McGuire, K.; Gerson, L.; Diokno, A. A 12 week, open label, phase I/IIa study using apatone for the treatment of prostate cancer patients who have failed standard therapy. Int. J. Med. Sci. 2008, 5, 62–67. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine Home Page, Double-Blinded Clinical Trial Using Apatone-B for Symptomatic Postoperative Total Joint Replacement (Apatone-B). ClinicalTrials.gov.Identifier: NCT01272830. Available online: https://clinicaltrials.gov (accessed on 20 April 2023).
- Hidalgo-Gutierres, A.; Gonzalez-Garcia, P.; Diaz-Casado, M.E.; Barriocanal-Casado, E.; Lopez-Herrador, S.; Quinzii, C.M.; Lopez, L.C. Metabolic targets of coenzyme Q10 in mitochondria. Antioxidants 2021, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Woerner, A.C.; Vockley, J. Mitochondrial disease and coenzyme Q10 deficiency: Commentary. J. Pediatr. 2021, 228, 14–15.E1. [Google Scholar] [CrossRef] [PubMed]
- Pallotti, F.; Bergamini, C.; Lamperti, C.; Fato, R. The role of coenzyme Q in disease: Direct and indirect involvement in cellular functions. Int. J. Mol. Sci. 2021, 23, 128. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P. Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell. Biol. 2014, 49, 105–111. [Google Scholar] [CrossRef]
- Bakalova, R.; Zhelev, Z.; Miller, T.; Aoki, I.; Higashi, T. New potential biomarker for stratification of patients for pharmacological vitamin C in adjuvant settings of cancer therapy. Redox. Biol. 2020, 28, 101357. [Google Scholar] [CrossRef]
- Pesini, A.; Hidalgo-Gutierrez, A.; Quinzii, C.M. Mechanisms and therapeutic effects of benzoquinone ring analogues in primary CoQ deficiency. Antioxidants 2022, 11, 665. [Google Scholar] [CrossRef]
- Ivanova, D.; Zhelev, Z.; Getsov, P.; Nikolova, B.; Aoki, I.; Higashi, T.; Bakalova, R. Vitamin K: Redox-modulation, prevention of mitochondrial dysfunction and anticancer effect. Redox. Biol. 2018, 16, 352–358. [Google Scholar] [CrossRef]
- Eleff, S.; Kennaway, N.G.; Buist, N.R.; Darley-Usmar, V.M.; Capaldi, R.A.; Bank, W.J.; Chance, B. 31P NMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle. Proc. Nat. Acad. Sci. USA 1984, 81, 3529–3533. [Google Scholar] [CrossRef]
- US Department of Health and Human Services (FDA) Home Page. National Institutes of Health (Office of Dietary Supplements). Dietary Supplements for Primary Mitochondrial Disorders. 2022. Available online: https://ods.od.nih.gov/factsheets/PrimaryMitochondrialDisorders-HealthProfessional/ (accessed on 20 October 2022).
- Hitomi, M.; Yokoyama, F.; Kita, Y.; Nonomura, T.; Masaki, T.; Yoshiji, H.; Inoue, H.; Kinekawa, F.; Kurokohchi, K.; Uchida, N.; et al. Antitumor effects of vitamins K1, K2 and K3 on hepatocellular carcinoma in vitro and in vivo. Int. J. Oncol. 2005, 26, 713–720. [Google Scholar] [CrossRef]
- Xu, W.; Wu, H.; Chen, S.; Wang, X.; Tanaka, S.; Sugiyama, K.; Yamada, H.; Hirano, T. Cytotoxic effects of vitamins K1, K2, K3 against human T lymphoblastoid leukemia cells through apoptosis induction and cell cycle arrest. Chem. Biol. Drug. Des. 2020, 96, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Kong, A. The therapeutic potential of imidazole or quinone-based compounds as radiosensitizers in combination with radiotherapy for the treatment of head and neck squamous cell carcinoma. Cancers 2022, 14, 4694. [Google Scholar] [CrossRef]
- Hirota, Y.; Tsugawa, N.; Nakagawa, K.; Suhara, Y.; Tanaka, K.; Uchino, Y.; Takeuchi, A.; Sawada, N.; Kamao, M.; Wada, A.; et al. Menadione (vitamin K3) is a catabolic product of oral phylloquinone (vitamin K1) in the intestine and a circulating precursor of tissue menaquinone-4 (vitamin 2). J. Biol. Chem. 2013, 288, 33071–33080. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Khan, I.A.; Shahbaz, M.; Qaisrani, T.B.; Fatmawati, S.; Abu-Izneid, T.; Imran, A.; Rahman, K.U.; Gondal, T.A. Thymoquinone: A novel strategy to combat cancer; a review. Biomed. Pharm. 2018, 106, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-L.; Tsai, C.-H.; Shrestha, S.; Lee, C.-C.; Liao, J.-W.; Hseu, Y.-C. Coenzyme Q0, a novel quinone derivative of Antrodia camphorata, induces ROS-mediated cytotoxic autophagy and apoptosis against human glioblastoma cells in vitro and in vivo. Food Chem. Toxicol. 2021, 155, 112384. [Google Scholar] [CrossRef]
- Mackei, M.; Voroshazi, J.; Sebok, C.; Neogrady, Z.; Matis, G.; Jerzsele, A. Fermented wheat germ extract as a redox modulator: Alleviating endotoxin-triggered oxidative stress in primary cultured rat hepatocytes. Oxid. Med. Cell. Longev. 2020, 2020, 3181202. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Voigt, W. Fermented wheat germ extract–nutritional supplement or anticancer drug? Nutr. J. 2011, 10, 89. [Google Scholar] [CrossRef]
- Graciani, F.S.; Ximenes, V.F. 2-Bromo-1,4-naphthoquinone: A potentially improved substitute of menadione in ApatoneTM therapy. Braz. J. Med. Biol. Res. 2012, 45, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Hirota, Y.; Sawada, N.; Yuge, N.; Watanabe, M.; Uchino, Y.; Okuda, N.; Shimomura, Y.; Suhara, Y.; Okano, T. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature 2010, 468, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.P.; Medana, C.; Stainier, D.Y.R.; et al. UBIAD1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas: UBIAD1 Protein Expression Summary. Available online: http://www.proteinatlas.org/ENSG00000120942-UBIAD1 (accessed on 20 October 2022).
- Li, H.; Qu, L.; Zhang, H.; Liu, J.; Zhang, X.A. Comprehensive transcriptomic landscape of cholangiosarcoma based on bioinformatics analysis from large cohort of patients. Sci. Rep. 2021, 11, 13713. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Chung, C.K.; Gonzalez, E.; Yoo, C. Causal inference network of genes relates with bone metastasis of breast cancer and osteoblasts using causal Bayesian networks. J. Bone Metab. 2018, 25, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wei, X.; Wu, S.; Wang, B.; Hong, L. Down-regulation of TERE1/UBIAD1 activated Ras-MAPK signaling and induced cell proliferations. Cell. Biol. Int. Rep. 2010, 17, e00005. [Google Scholar] [CrossRef] [PubMed]
- Frederiks, W.J.; Sepulveda, J.; Lai, P.; Tomaszewski, J.E.; Lin, M.-F.; McGarvey, T.; Rauscher, F.J.; Malkovicz, S.B. The tumor suppressor TERE1 (UBIAD1) prenyltransferase regulates the elevated cholesterol phenotype in castration resistant prostate cancer by controlling a program of ligand dependent SXR target genes. Oncotarget 2013, 4, 1075–1092. [Google Scholar] [CrossRef]
- Greenwood, J.; Steinman, L.; Zamvil, R. Statin therapy in autoimmunity: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 350–370. [Google Scholar] [CrossRef] [PubMed]
- Esposti, M.D.; Ngo, A.; McMullen, G.L.; Ghelli, A.; Sparla, F.; Benelli, B.; Ratta, M.; Linnane, A.W. The specificity of mitochondrial complex I for ubiqu1inones. Biochem. J. 1996, 313, 327–334. [Google Scholar] [CrossRef]
- Lenaz, G. Quinone specificity of complex I. Biochim. Biophys. Acta 1998, 1364, 207–221. [Google Scholar] [CrossRef]
- Cerqua, C.; Casarin, A.; Pierrel, F.; Fonseca, L.V.; Viola, G.; Salviati, L.; Trevisson, E. Vitamin K2 cannot substitute coenzyme Q10 as electron carrier in the mitochondrial respiratory chain of mammalian cells. Sci. Rep. 2019, 9, 6553. [Google Scholar] [CrossRef]
- Erb, M.; Hoffmann-Enger, B.; Deppe, H.; Soeberdt, M.; Haefeli, R.H.; Rummey, C.; Feurer, A.; Gueven, N. Features of idebenone and related short-chain quinones that rescue ATP levels under conditions of impaired mitochondrial complex I. PLoS ONE 2012, 7, e36153. [Google Scholar] [CrossRef]
- Zhang, H.; Osyczka, A.; Dutton, P.L.; Moser, C.C. Exposing the complex III Qo semiquinone radical. Biochim. Biophys. Acta 2007, 1767, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Okayashi, H.; Ishihara, M.; Satoh, K.; Sakagami, H. Cytotoxic activity of vitamins K1, K2 and K3 against human oral tumor cell lines. Anticancer Res. 2001, 21, 2387–2392. [Google Scholar]
- Njus, D.; Asmaro, K.; Li, G.; Palomino, E. Redox cycling of quinones reduced by ascorbic acid. Chem. Biol. Interact. 2023, 373, 110397. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.P.; Karandrea, S.; Burgos, D.Z.; Jaiswal, A.A.; Heart, E.A. NAD(P)H-dependent quinone oxidoreductase 1 (NQO1) and cytochrome P450 oxidoreductase (CYP450OR) differentially regulate menadione-mediated alterations in redox status, survival, metabolism in pancreatic beta-cells. Toxicol. Lett. 2016, 262, 1–11. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H: Quinone oxidoreductase 1 (NQO1) as a therapeutic and diagnostic target in cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef]
- Nikiforova, A.B.; Saris, N.-E.L.; Kruglov, A.G. External mitochondrial NADH-dependent reductase of redox cyclers: VDAC1 and Cyb5R3? Free Radic. Biol. Med. 2014, 74, 74–84. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Guo, S.; Wang, G. Glutathione peroxidases as oncotargets. Oncotarget 2017, 8, 80093–80102. [Google Scholar] [CrossRef]
- Alharbi, Y.; Kapur, A.; Felder, M.; Barroilhet, L.; Pattnaik, B.R.; Patankar, M.S. Oxidative stress induced by the anti-cancer agents, plumbagin, atovaquone, inhibits ion transport through Na+/K+-ATPase. Sci. Rep. 2020, 10, 19585. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Frezza, C.; Erez, A. Targeting metabolic plasticity and flexibility dynamics for cancer therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
- Verrax, J.; Vanbever, S.; Stockis, J.; Taper, H.; Calderon, P.B. Role of glycolysis inhibition and poly-(ADP-ribose) polymerase activation in necrotic-like cell death caused by ascorbate/menadione-induced oxidative stress in K562 human chronic myelogenous leukemic cells. Int. J. Cancer 2007, 120, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Tsai, T.-J.; Korivi, M.; Liu, J.-Y.; Chen, H.-J.; Lin, C.-M.; Shen, Y.-C.; Yang, H.-L. Antitumor properties of coenzyme Q0 against human ovarian carcinoma cells via induction of ROS-mediated apoptosis and cytoprotective autophagy. Sci. Rep. 2017, 7, 8062. [Google Scholar] [CrossRef] [PubMed]
- Despotovic, A.; Mircic, A.; Misirlic-Dencic, S.; Harhaji-Trajkovic, L.; Trajkovic, V.; Zogovic, N.; Tovilovic-Kovacevic, G. Combination of ascorbic acid and menadione induces cytotoxic autophagy in human glioblastoma cells. Oxid. Med. Cell. Longev. 2022, 2022, 2998132. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Suzuki, Y.; Yonezawa, Y.; Ota, Y.; Okamoto, Y.; Demizu, Y.; Huang, P.; Yoshida, H.; Sugimura, K.; Mizushina, Y. DNA polymerase gamma inhibition by vitamin K3 induces mitochondria-mediated cytotoxicity in human cancer cells. Cancer Sci. 2008, 99, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, Y.; Kuriyama, I.; Fukuoh, A.; Muta, T.; Kang, D.; Takemura, M.; Kato, I.; Yoshida, H.; Mizushima, Y. Inhibitory effect of coenzyme Q1 on eukaryitic DNA polymerase gamma and DNA topoisomerase II activities on the growth of a human cancer cell line. Cancer Sci. 2006, 97, 716–723. [Google Scholar] [CrossRef]
- Hseu, Y.-C.; Thiyagarajan, V.; Ou, T.-T.; Yang, H.-L. CoQ0-induced mitochondrial PTP opening triggers apoptosis via ROS-mediated VDAC1 upregulation in HL-2 leukemia cells and suppresses tumor growth in athymic nude mice/xenografted nude mice. Arch. Toxicol. 2018, 92, 301–322. [Google Scholar] [CrossRef]
- Cheng, H.-L.; Lee, Y.-H.; Yuan, T.-M.; Chen, S.-W.; Chueh, P.-J. Update on a tumor-associated NADH oxidase in gastric cancer cell growth. World J. Gastroenterol. 2016, 22, 2900–2905. [Google Scholar] [CrossRef]
- Jiang, Z.; Gorenstein, N.M.; Morré, D.M.; Morré, D.J. Molecular cloning and characterization of a candidate human growth-related and time-keeping constitutive cell surface hydroquinone (NADH) oxidase. Biochemistry 2008, 47, 14028. [Google Scholar] [CrossRef]
- Su, Y.-C.; Lin, Y.-H.; Zeng, Z.-M.; Shao, K.-N.; Chueh, P.J. Chemotherapeutic agents enhance cell migration and epithelial-to-mesenchymal transition through transient up-regulation of tNOX (ENOX2) protein. Biochim. Biophys. Acta 2012, 1820, 1744–1752. [Google Scholar] [CrossRef]
- Liu, N.-C.; Hsieh, P.-F.; Hsieh, M.-K.; Zeng, Z.-M.; Cheng, H.-L.; Liao, J.-W.; Chueh, P.J. Capsaicin-mediated tNOX (ENOX2) up-regulation enhances cell proliferation and migration in vitro and in vivo. J. Agric. Food Chem. 2012, 60, 2758–2765. [Google Scholar] [CrossRef]
- The Human Protein Atlas: ENOX2 Protein Expression Summary. Available online: http://www.proteinatlas.org/ENSG00000165675-ENOX2 (accessed on 17 November 2022).
- Islam, A.; Hsieh, P.-F.; Liu, P.-F.; Chou, J.-C.; Liao, J.-W.; Hsieh, M.-K.; Chueh, P.J. Capsaicin exerts therapeutic effects by targeting tNOX-SIRT1 axis and augmenting ROS-dependent cytotoxic autophagy in melanoma cancer cells. Am. J. Cancer Res. 2021, 11, 4199–4219. [Google Scholar] [PubMed]
- Tang, X.; Tian, Z.; Chueh, P.-J.; Chen, S.; Morre, D.M.; Morre, D.J. Alternative splicing as the basis for specific localization of tNOX, a unique hydroquinone (NADH) oxidase to the cancer cell surface. Biochemistry 2007, 46, 12337–12346. [Google Scholar] [CrossRef] [PubMed]
- Gilloteaux, J.; Jamison, J.M.; Arnold, D.; Taper, H.S.; Summers, J.L. Ultrastructural aspects of autoschizis: A new cancer cell death induced by the synergistic action of ascorbate/menadione on human bladder carcinoma cells. Ultrastruct. Pathol. 2001, 25, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hou, Y.; He, H.; Chen, Y.; Zhou, R.; Wang, X.; Gong, T.; Jiang, W. Synthetic vitamin K analogs inhibit inflammation by targeting the NLRP3 inflammasome. Cell. Mol. Immunol. 2021, 18, 2422–2430. [Google Scholar] [CrossRef]
- Hseu, Y.-C.; Tseng, Y.-F.; Pandey, S.; Shrestha, S.; Lin, K.-Y.; Lin, C.-W.; Lee, C.-C.; Huang, S.-T.; Yang, H.-L. Coenzyme Q0 inhibits NLRP3 inflammasome activation through mitophagy induction in LPS/ATP-stimulated macrophages. Oxid. Med. Cell. Longev. 2022, 2022, 4266214. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox. Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakalova, R.; Lazarova, D.; Sumiyoshi, A.; Shibata, S.; Zhelev, Z.; Nikolova, B.; Semkova, S.; Vlaykova, T.; Aoki, I.; Higashi, T. Redox-Cycling “Mitocans” as Effective New Developments in Anticancer Therapy. Int. J. Mol. Sci. 2023, 24, 8435. https://doi.org/10.3390/ijms24098435
Bakalova R, Lazarova D, Sumiyoshi A, Shibata S, Zhelev Z, Nikolova B, Semkova S, Vlaykova T, Aoki I, Higashi T. Redox-Cycling “Mitocans” as Effective New Developments in Anticancer Therapy. International Journal of Molecular Sciences. 2023; 24(9):8435. https://doi.org/10.3390/ijms24098435
Chicago/Turabian StyleBakalova, Rumiana, Dessislava Lazarova, Akira Sumiyoshi, Sayaka Shibata, Zhivko Zhelev, Biliana Nikolova, Severina Semkova, Tatyana Vlaykova, Ichio Aoki, and Tatsuya Higashi. 2023. "Redox-Cycling “Mitocans” as Effective New Developments in Anticancer Therapy" International Journal of Molecular Sciences 24, no. 9: 8435. https://doi.org/10.3390/ijms24098435
APA StyleBakalova, R., Lazarova, D., Sumiyoshi, A., Shibata, S., Zhelev, Z., Nikolova, B., Semkova, S., Vlaykova, T., Aoki, I., & Higashi, T. (2023). Redox-Cycling “Mitocans” as Effective New Developments in Anticancer Therapy. International Journal of Molecular Sciences, 24(9), 8435. https://doi.org/10.3390/ijms24098435