

The Discovery and Characterization of Conserved and Novel miRNAs in the Different Developmental Stages and Organs of Pikeperch (Sander lucioperca)

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification of Conserved and Novel miRNA Genes in Pikeperch
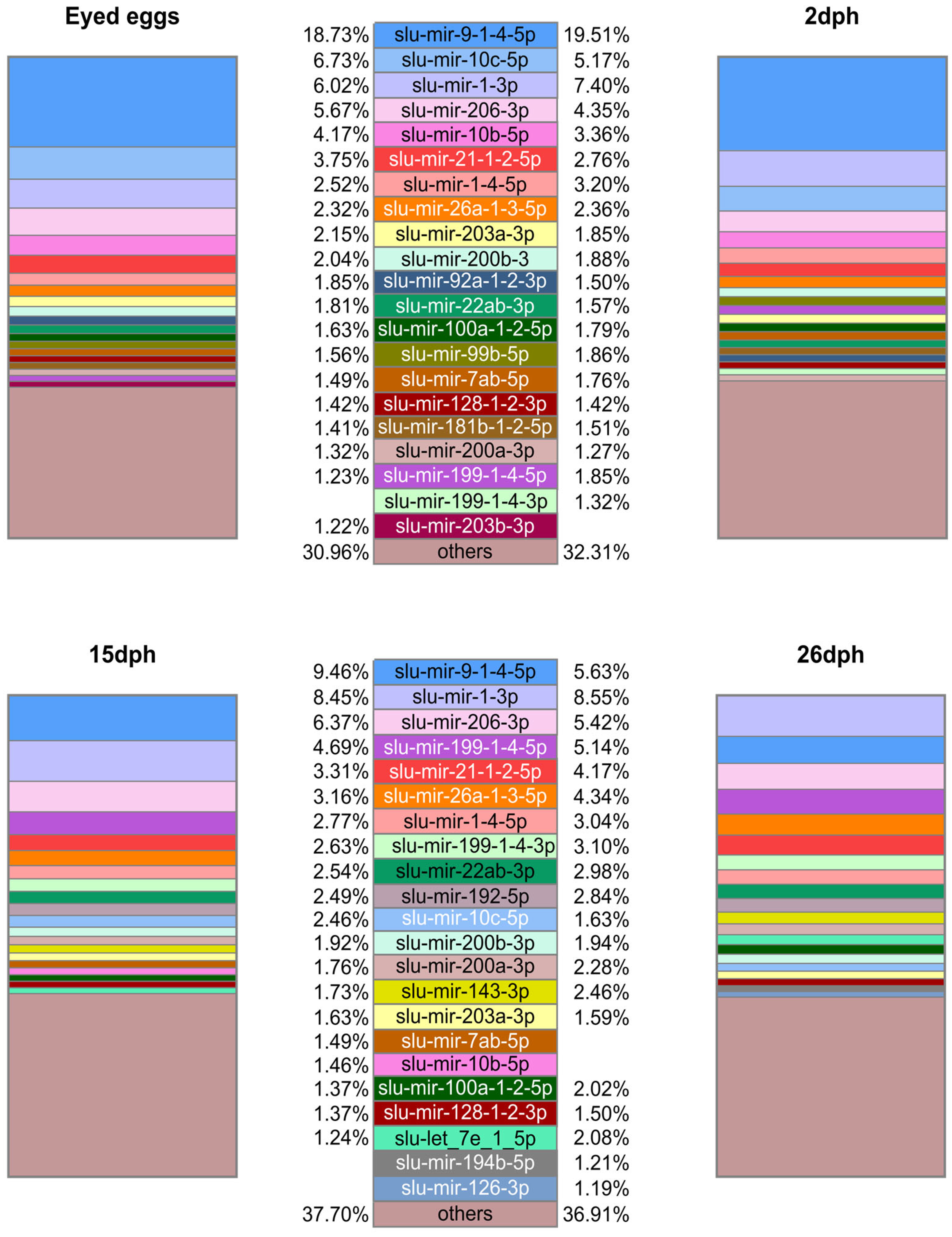
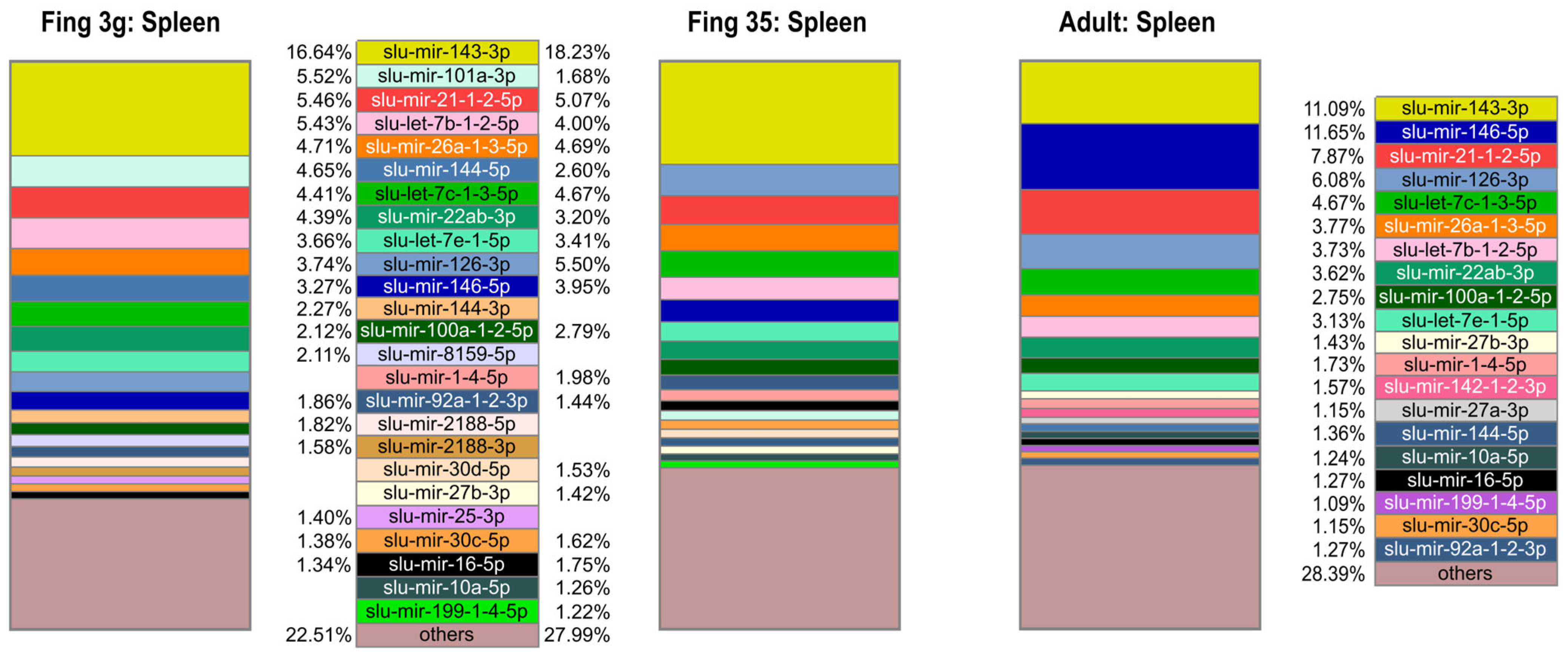
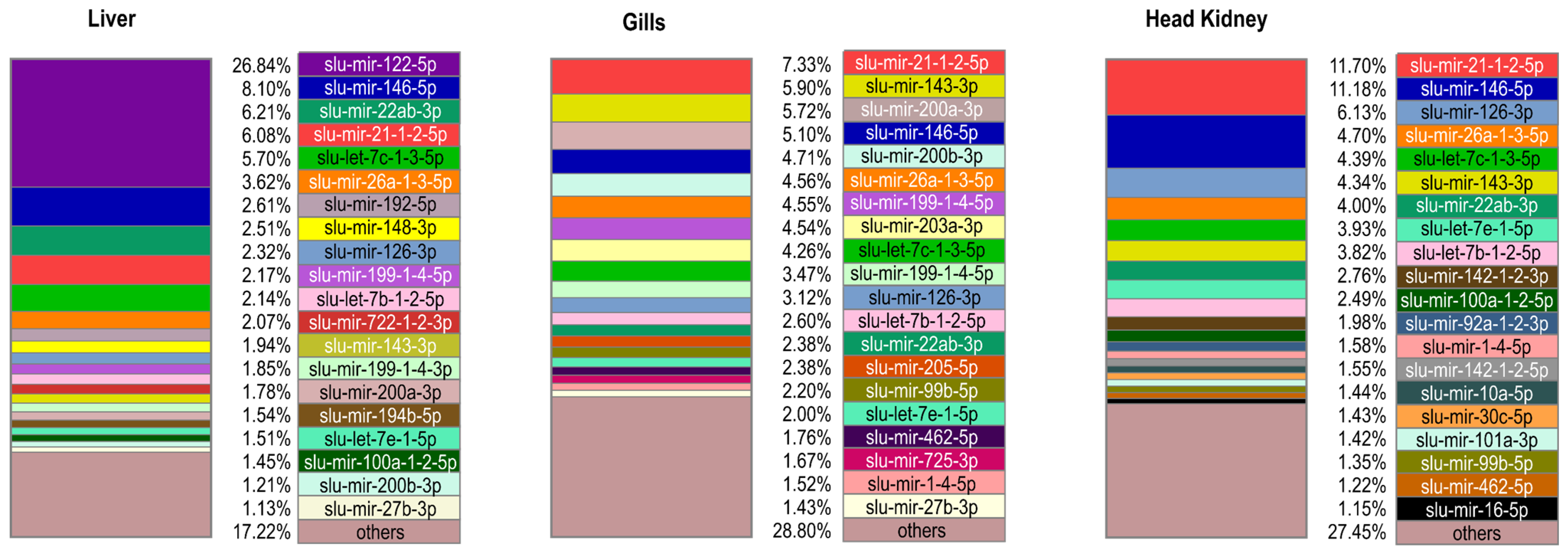
2.2. Mature miRNA Diversity across Sample Groups and Their Relative Abundance within Sample Groups
2.3. Differential Expression Analysis and qPCR Revealed Additional miRNAs Associated with Particular Life Stages and Organs
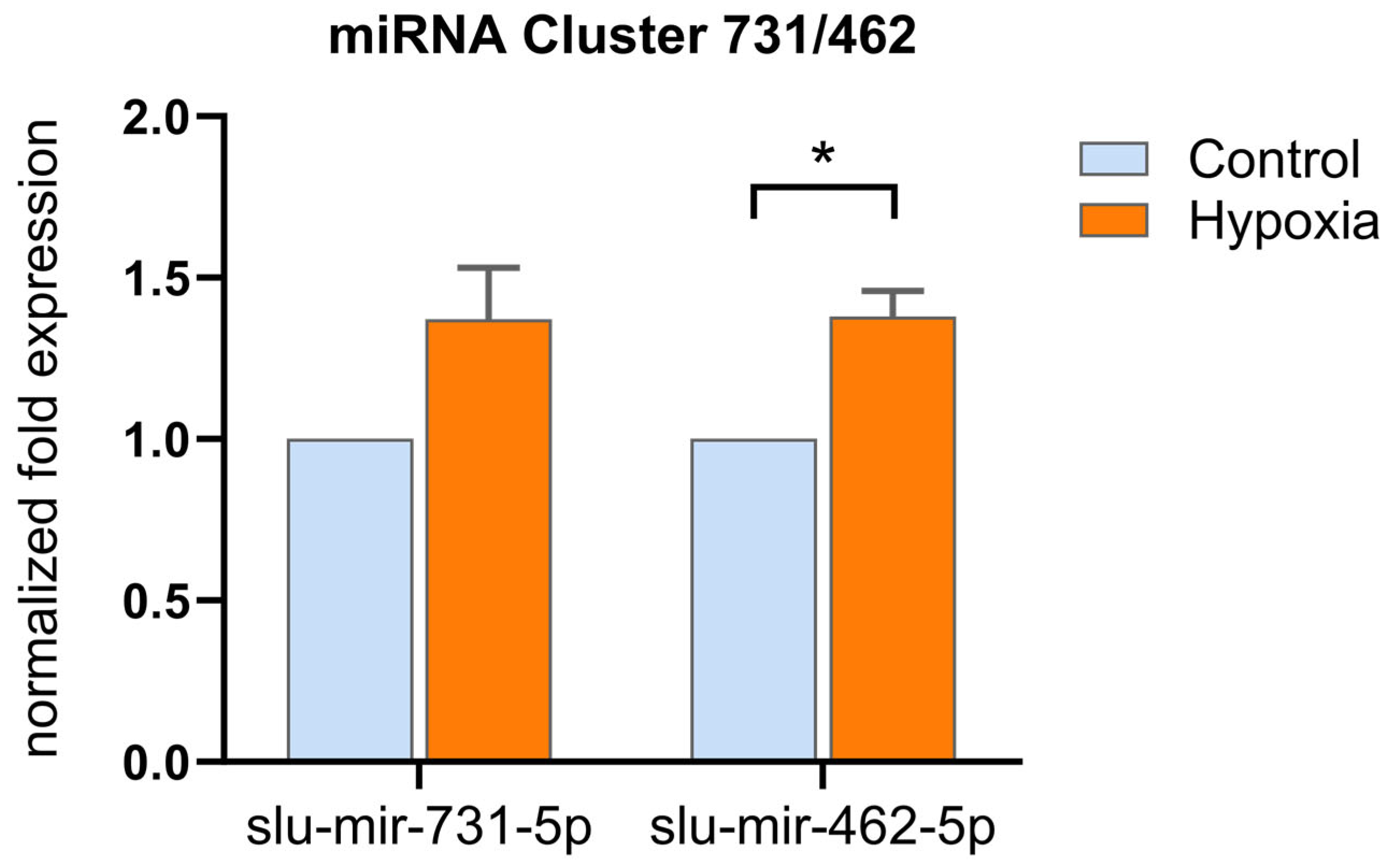
2.4. The miR-462/miR-731 Cluster Is Associated with Hypoxia Stress in Pikeperch
3. Materials and Methods
3.1. Fish Sample Material
3.2. Small RNA Isolation, Library Preparation, and Sequencing (Ilumina Technology)
3.3. Pre-Processing and miRDeep2 Analysis of Sequencing Data
3.4. Identification of miRNA Gene Clusters
3.5. Prediction of Enriched and Putative Differently Expressed (DE) miRNAs in and among Developmental Stages and Organs
3.6. Stability Testing of Possible Reference miRNAs and RT-qPCR Analysis
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.F.; Pasquinelli, A.E. MicroRNA biogenesis: Regulating the regulators. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Stefani, G.; Slack, F.J. Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell Biol. 2008, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Ståhle, M.; Pivarcsi, A. MicroRNAs and immunity: Novel players in the regulation of normal immune function and inflammation. Semin. Cancer Biol. 2008, 18, 131–140. [Google Scholar] [CrossRef]
- Ramberg, S.; Andreassen, R. MicroSalmon: A Comprehensive, Searchable Resource of Predicted MicroRNA Targets and 3′UTR Cis-Regulatory Elements in the Full-Length Sequenced Atlantic Salmon Transcriptome. Non-Coding RNA 2021, 7, 61. [Google Scholar] [CrossRef]
- Bizuayehu, T.T.; Babiak, I. MicroRNA in Teleost Fish. Genome Biol. Evol. 2014, 6, 1911–1937. [Google Scholar] [CrossRef]
- Andreassen, R.; Høyheim, B. miRNAs associated with immune response in teleost fish. Dev. Comp. Immunol. 2017, 75, 77–85. [Google Scholar] [CrossRef]
- Herkenhoff, M.E.; Oliveira, A.C.; Nachtigall, P.G.; Costa, J.M.; Campos, V.F.; Hilsdorf, A.W.S.; Pinhal, D. Fishing into the MicroRNA Transcriptome. Front. Genet. 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Presslauer, C.; Tilahun Bizuayehu, T.; Kopp, M.; Fernandes, J.M.O.; Babiak, I. Dynamics of miRNA transcriptome during gonadal development of zebrafish. Sci. Rep. 2017, 7, 43850. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Manninga, H.; Slanchev, K.; Chien, M.; Russo, J.J.; Ju, J.; Sheridan, R.; John, B.; Marks, D.S.; Gaidatzis, D.; et al. The developmental miRNA profiles of zebrafish as determined by small RNA cloning. Genes Dev. 2005, 19, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Woldemariam, N.T.; Agafonov, O.; Høyheim, B.; Houston, R.D.; Taggart, J.B.; Andreassen, R. Expanding the miRNA Repertoire in Atlantic Salmon; Discovery of IsomiRs and miRNAs Highly Expressed in Different Tissues and Developmental Stages. Cells 2019, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Shwe, A.; Krasnov, A.; Visnovska, T.; Ramberg, S.; Østbye, T.-K.K.; Andreassen, R. Differential Expression of miRNAs and Their Predicted Target Genes Indicates That Gene Expression in Atlantic Salmon Gill Is Post-Transcriptionally Regulated by miRNAs in the Parr-Smolt Transformation and Adaptation to Sea Water. Int. J. Mol. Sci. 2022, 23, 8831. [Google Scholar] [CrossRef] [PubMed]
- Juanchich, A.; Bardou, P.; Rué, O.; Gabillard, J.-C.; Gaspin, C.; Bobe, J.; Guiguen, Y. Characterization of an extensive rainbow trout miRNA transcriptome by next generation sequencing. BMC Genom. 2016, 17, 164. [Google Scholar] [CrossRef]
- Cardona, E.; Guyomar, C.; Desvignes, T.; Montfort, J.; Guendouz, S.; Postlethwait, J.H.; Skiba-Cassy, S.; Bobe, J. Circulating miRNA repertoire as a biomarker of metabolic and reproductive states in rainbow trout. BMC Biol. 2021, 19, 235. [Google Scholar] [CrossRef]
- Huang, C.W.; Li, Y.H.; Hu, S.Y.; Chi, J.R.; Lin, G.H.; Lin, C.C.; Gong, H.Y.; Chen, J.Y.; Chen, R.H.; Chang, S.J.; et al. Differential expression patterns of growth-related microRNAs in the skeletal muscle of Nile tilapia (Oreochromis niloticus). J. Anim. Sci. 2012, 90, 4266–4279. [Google Scholar] [CrossRef]
- Herkenhoff, M.E.; Bovolenta, L.A.; Broedel, O.; Dos Santos, L.D.; de Oliveira, A.C.; Chuffa, L.G.A.; Ribeiro, A.d.O.; Lupi, L.A.; Dias, M.A.D.; Hilsdorf, A.W.S.; et al. Variant expression signatures of microRNAs and protein related to growth in a crossbreed between two strains of Nile tilapia (Oreochromis niloticus). Genomics 2021, 113, 4303–4312. [Google Scholar] [CrossRef]
- Eslamloo, K.; Inkpen, S.M.; Rise, M.L.; Andreassen, R. Discovery of microRNAs associated with the antiviral immune response of Atlantic cod macrophages. Mol. Immunol. 2018, 93, 152–161. [Google Scholar] [CrossRef]
- Andreassen, R.; Rangnes, F.; Sivertsen, M.; Chiang, M.; Tran, M.; Worren, M.M. Discovery of miRNAs and Their Corresponding miRNA Genes in Atlantic Cod (Gadus morhua): Use of Stable miRNAs as Reference Genes Reveals Subgroups of miRNAs That Are Highly Expressed in Particular Organs. PLoS ONE 2016, 11, e0153324. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Woldemariam, N.T.; Visnovska, T.; Rise, M.L.; Boyce, D.; Santander, J.; Andreassen, R. Characterization of miRNAs in Embryonic, Larval, and Adult Lumpfish Provides a Reference miRNAome for Cyclopterus lumpus. Biology 2022, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, J.; Li, X.; Ge, J.; Pan, J.; Xu, X. Identification and Characterization of MicroRNAs in Channel Catfish (Ictalurus punctatus) by Using Solexa Sequencing Technology. PLoS ONE 2013, 8, e54174. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Fu, J.; Tan, X.; Shi, Y.; Ye, J.; Guan, W.; Shi, Y.; Xu, M. The miR-155 regulates cytokines expression by SOSC1 signal pathways of fish in vitro and in vivo. Fish Shellfish. Immunol. 2020, 106, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Bardou, P.; Montfort, J.; Sydes, J.; Guyomar, C.; George, S.; Postlethwait, J.H.; Bobe, J. FishmiRNA: An Evolutionarily Supported MicroRNA Annotation and Expression Database for Ray-Finned Fishes. Mol. Biol. Evol. 2022, 39, msac004. [Google Scholar] [CrossRef] [PubMed]
- Woldemariam, N.T.; Agafonov, O.; Sindre, H.; Høyheim, B.; Houston, R.D.; Robledo, D.; Bron, J.E.; Andreassen, R. miRNAs Predicted to Regulate Host Anti-viral Gene Pathways in IPNV-Challenged Atlantic Salmon Fry Are Affected by Viral Load, and Associated with the Major IPN Resistance QTL Genotypes in Late Infection. Front. Immunol. 2020, 11, 2113. [Google Scholar] [CrossRef] [PubMed]
- Ramberg, S.; Krasnov, A.; Colquhoun, D.; Wallace, C.; Andreassen, R. Expression Analysis of Moritella viscosa-Challenged Atlantic Salmon Identifies Disease-Responding Genes, MicroRNAs and Their Predicted Target Genes and Pathways. Int. J. Mol. Sci. 2022, 23, 11200. [Google Scholar] [CrossRef] [PubMed]
- Wienholds, E.; Plasterk, R.H.A. MicroRNA function in animal development. FEBS Lett. 2005, 579, 5911–5922. [Google Scholar] [CrossRef]
- Su, H.; Fan, J.; Ma, D.; Zhu, H. Identification and Characterization of Osmoregulation Related MicroRNAs in Gills of Hybrid Tilapia under Three Types of Osmotic Stress. Front. Genet. 2021, 12, 526277. [Google Scholar] [CrossRef]
- Huang, C.-H.; Chen, N.; Huang, C.-X.; Zhang, B.; Wu, M.; He, L.; Liu, H.; Tang, R.; Wang, W.-M.; Wang, H.-L. Involvement of the miR-462/731 cluster in hypoxia response in Megalobrama amblycephala. Fish Physiol. Biochem. 2017, 43, 863–873. [Google Scholar] [CrossRef]
- Østbye, T.-K.K.; Woldemariam, N.T.; Lundberg, C.E.; Berge, G.M.; Ruyter, B.; Andreassen, R. Modulation of hepatic miRNA expression in Atlantic salmon (Salmo salar) by family background and dietary fatty acid composition. J. Fish Biol. 2021, 98, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- FAO. In Brief to the State of World Fisheries and Aquaculture 2022; FAO: Rome, Italy, 2022. [Google Scholar]
- FAO. The State of World Fisheries and Aquaculture 2020. Sustainability in Action; FAO: Rome, Italy, 2020. [Google Scholar]
- Jankowska, B.; Zakes, Z.; Zmijewski, T.; Szczepkowski, M. A comparison of selected quality features of the tissue and slaughter yield of wild and cultivated pikeperch Sander lucioperca (L.). Eur. Food Res. Technol. 2003, 217, 401–405. [Google Scholar] [CrossRef]
- Kestemont, P.; Dabrowski, K.; Summerfelt, R.C. Biology and Culture of Percid Fishes: Principles and Practices; Springer: Dordrecht, The Netherlands, 2015; ISBN 9789401772273. [Google Scholar]
- Zakęś, Z. FAO 2023 Cultured Aquatic Species Information Programme. Sander lucioperca; FAO: Rome, Italy, 2012. [Google Scholar]
- Fontaine, P.; Kestemont, P.; Teletchea, F.; Wang, N. Percid Fish Culture. From Research to Production; Presses Universitaires de Namur: Namur, Belgium, 2008; ISBN 9782870375822. [Google Scholar]
- Policar, T.; Schaefer, F.J.; Panana, E.; Meyer, S.; Teerlinck, S.; Toner, D.; Żarski, D. Recent progress in European percid fish culture production technology—Tackling bottlenecks. Aquac. Int. 2019, 27, 1151–1174. [Google Scholar] [CrossRef]
- Steenfeldt, S. Chapter 10: Culture Methods of Pikeperch Early Life Stages. In Biology and Culture of Percid Fishes; Springer: Dordrecht, The Netherlands, 2015; pp. 295–312. ISBN 9789401772273. [Google Scholar]
- Liu, Y.; Rosten, T.W.; Henriksen, K.; Hognes, E.S.; Summerfelt, S.; Vinci, B. Comparative economic performance and carbon footprint of two farming models for producing Atlantic salmon (Salmo salar): Land-based closed containment system in freshwater and open net pen in seawater. Aquac. Eng. 2016, 71, 1–12. [Google Scholar] [CrossRef]
- Bregnballe, J.; Eurofish; FAO. A Guide to Recirculation Aquaculture: An Introduction to the New Environmentally Friendly and Highly Productive Closed Fish Farming Systems; Food and Agriculture Organization of the United Nations: Rome, Italy, 2022; ISBN 9789251369814. [Google Scholar]
- Perry, W.B.; Solberg, M.F.; Besnier, F.; Dyrhovden, L.; Matre, I.H.; Fjelldal, P.G.; Ayllon, F.; Creer, S.; Llewellyn, M.; Taylor, M.I.; et al. Evolutionary drivers of kype size in Atlantic salmon (Salmo salar): Domestication, age and genetics. R. Soc. Open Sci. 2019, 6, 190021. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, S.J.; Kess, T.; Bentzen, P.; Kent, M.P.; Lien, S.; Gilbey, J.; Clément, M.; Jeffery, N.W.; Waples, R.S.; Bradbury, I.R. Genomic signatures and correlates of widespread population declines in salmon. Nat. Commun. 2019, 10, 2996. [Google Scholar] [CrossRef] [PubMed]
- García-Pérez, I.; Molsosa-Solanas, A.; Perelló-Amorós, M.; Sarropoulou, E.; Blasco, J.; Gutiérrez, J.; Garcia de la Serrana, D. The Emerging Role of Long Non-Coding RNAs in Development and Function of Gilthead Sea Bream (Sparus aurata) Fast Skeletal Muscle. Cells 2022, 11, 428. [Google Scholar] [CrossRef] [PubMed]
- Nguinkal, J.A.; Brunner, R.M.; Verleih, M.; Rebl, A.; de Los Ríos-Pérez, L.; Schäfer, N.; Hadlich, F.; Stüeken, M.; Wittenburg, D.; Goldammer, T. The First Highly Contiguous Genome Assembly of Pikeperch (Sander lucioperca), an Emerging Aquaculture Species in Europe. Genes 2019, 10, 708. [Google Scholar] [CrossRef]
- de Los Ríos-Pérez, L.; Nguinkal, J.A.; Verleih, M.; Rebl, A.; Brunner, R.M.; Klosa, J.; Schäfer, N.; Stüeken, M.; Goldammer, T.; Wittenburg, D. An ultra-high density SNP-based linkage map for enhancing the pikeperch (Sander lucioperca) genome assembly to chromosome-scale. Sci. Rep. 2020, 10, 22335. [Google Scholar] [CrossRef]
- Nguinkal, J.A.; Verleih, M.; De Los Ríos-Pérez, L.; Brunner, R.M.; Sahm, A.; Bej, S.; Rebl, A.; Goldammer, T. Comprehensive Characterization of Multitissue Expression Landscape, Co-Expression Networks and Positive Selection in Pikeperch. Cells 2021, 10, 2289. [Google Scholar] [CrossRef]
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005, 33, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-C.; Chan, W.-C.; Hu, L.-Y.; Lai, C.-H.; Hsu, C.-N.; Lin, W. Identification of homologous microRNAs in 56 animal genomes. Genom. 2010, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Koonin, E.V. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 2008, 23, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shen, Y.; Wu, Q.; Kumar, S.; He, B.; Shi, S.; Carthew, R.W.; Wang, S.M.; Wu, C.-I. The birth and death of microRNA genes in Drosophila. Nat. Genet. 2008, 40, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Baskerville, S.; Bartel, D.P. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Lagos-quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Giraldez, A.J.; Mishima, Y.; Rihel, J.; Grocock, R.J.; Van Dongen, S.; Inoue, K.; Enright, A.J.; Schier, A.F. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science 2006, 312, 75–79. [Google Scholar] [CrossRef]
- Giusti, J.; Pinhal, D.; Moxon, S.; Campos, C.L.; Münsterberg, A.; Martins, C. MicroRNA-10 modulates Hox genes expression during Nile tilapia embryonic development. Mech. Dev. 2016, 140, 12–18. [Google Scholar] [CrossRef]
- Tehler, D.; Høyland-Kroghsbo, N.M.; Lund, A.H. The miR-10 microRNA precursor family. RNA Biol. 2011, 8, 728–734. [Google Scholar] [CrossRef]
- Radhakrishnan, B.; Alwin Prem Anand, A. Role of miRNA-9 in Brain Development. J. Exp. Neurosci. 2016, 10, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Tessmar-Raible, K.; Raible, F.; Christodoulou, F.; Guy, K.; Rembold, M.; Hausen, H.; Arendt, D. Conserved sensory-neurosecretory cell types in annelid and fish forebrain: Insights into hypothalamus evolution. Cell 2007, 129, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, A.J.; Cinalli, R.M.; Glasner, M.E.; Enright, A.J.; Thomson, J.M.; Baskerville, S.; Hammond, S.M.; Bartel, D.P.; Schier, A.F. MicroRNAs regulate brain morphogenesis in zebrafish. Science 2005, 308, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cassidy, J.J.; Reinke, C.A.; Fischboeck, S.; Carthew, R.W. A microRNA imparts robustness against environmental fluctuation during development. Cell 2009, 137, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Mitchelson, K.R.; Qin, W.-Y. Roles of the canonical myomiRs miR-1, -133 and -206 in cell development and disease. World J. Biol. Chem. 2015, 6, 162–208. [Google Scholar] [CrossRef] [PubMed]
- Townley-Tilson, W.H.D.; Callis, T.E.; Wang, D. MicroRNAs 1, 133, and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. Int. J. Biochem. Cell Biol. 2010, 42, 1252–1255. [Google Scholar] [CrossRef]
- Esau, C.; Kang, X.; Peralta, E.; Hanson, E.; Marcusson, E.G.; Ravichandran, L.V.; Sun, Y.; Koo, S.; Perera, R.J.; Jain, R.; et al. MicroRNA-143 Regulates Adipocyte Differentiation. J. Biol. Chem. 2004, 279, 52361–52365. [Google Scholar] [CrossRef]
- Mennigen, J.A.; Skiba-Cassy, S.; Panserat, S. Ontogenesis of expression of metabolic genes and microRNAs in rainbow trout alevins during the transition from the endogenous to the exogenous feeding period. J. Exp. Biol. 2013, 216, 1597–1608. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Simmini, S.; Abreu-Goodger, C.; Bartonicek, N.; Di Giacomo, M.; Bilbao-Cortes, D.; Horos, R.; Von Lindern, M.; Enright, A.J.; O’carroll, D. The miR-144/451 locus is required for erythroid homeostasis. J. Exp. Med. 2010, 207, 1351–1358. [Google Scholar] [CrossRef]
- Zhao, Y.; Fan, S.; Yuan, P.; Li, G. Expression characteristics and interaction networks of microRNAs in spleen tissues of grass carp (Ctenopharyngodon idella). PLoS ONE 2022, 17, e0266189. [Google Scholar] [CrossRef]
- Cui, J.; Gao, Y.; Chu, Q.; Bi, D.; Xu, T. miRNA-8159 is involved in TLR signaling pathway regulation after pathogen infection by direct targeting TLR13 in miiuy croaker. Fish Shellfish. Immunol. 2017, 66, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2012, 9, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.-J.; Yao, Y.; Cai, X.-Y.; Fang, G.-Y. Emerging Role of MiR-192-5p in Human Diseases. Front. Pharmacol. 2021, 12, 614068. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.M.; Ho, J.C.H.; Nong, W.; Hui, J.H.L.; Lai, K.P.; Wong, C.K.C. Genome-wide analysis of MicroRNA-messenger RNA interactome in ex-vivo gill filaments, Anguilla japonica. BMC Genom. 2020, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Johansen, I.; Andreassen, R. Validation of miRNA genes suitable as reference genes in qPCR analyses of miRNA gene expression in Atlantic salmon (Salmo salar). BMC Res. Notes 2014, 8, 945. [Google Scholar] [CrossRef]
- Kunz, Y.W. Developmental Biology of Teleost Fishes; Springer: Dordrecht, The Netherlands, 2004; ISBN 9781402029974. [Google Scholar]
- Alberti, C.; Cochella, L. A framework for understanding the roles of miRNAs in animal development. Development 2017, 144, 2548–2559. [Google Scholar] [CrossRef]
- Foshay, K.M.; Gallicano, G.I. miR-17 family miRNAs are expressed during early mammalian development and regulate stem cell differentiation. Dev. Biol. 2009, 326, 431–443. [Google Scholar] [CrossRef]
- Singh, S.; Saini, H.; Sharma, A.; Gupta, S.; Huddar, V.G.; Tripathi, R. Breast cancer: miRNAs monitoring chemoresistance and systemic therapy. Front. Oncol. 2023, 13, 1155254. [Google Scholar] [CrossRef]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The microRNA-17-92 family of microRNA clusters in development and disease. Cancer J. 2012, 18, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.L.; Alvarez-Saavedra, E.; Miska, E.A.; Lau, N.C.; Bartel, D.P.; Horvitz, H.R.; Ambros, V. The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev. Cell 2005, 9, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Batzel, P.; Sydes, J.; Eames, B.F.; Postlethwait, J.H. miRNA analysis with Prost! reveals evolutionary conservation of organ-enriched expression and post-transcriptional modifications in three-spined stickleback and zebrafish. Sci. Rep. 2019, 9, 3913. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.; Sundaram, A.Y.; Valente, L.M.; Conceição, L.E.; Engrola, S.; Fernandes, J.M. Thermal plasticity of the miRNA transcriptome during Senegalese sole development. BMC Genom. 2014, 15, 525. [Google Scholar] [CrossRef] [PubMed]
- Richbourg, H.A.; Hu, D.P.; Xu, Y.; Barczak, A.J.; Marcucio, R.S. miR-199 family contributes to regulation of sonic hedgehog expression during craniofacial development. Dev. Dyn. 2020, 249, 1062–1076. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Contreras, A.; Postlethwait, J.H. Evolution of the miR199-214 cluster and vertebrate skeletal development. RNA Biol. 2014, 11, 281–294. [Google Scholar] [CrossRef]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.-H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef]
- Vacante, F.; Denby, L.; Sluimer, J.C.; Baker, A.H. The function of miR-143, miR-145 and the MiR-143 host gene in cardiovascular development and disease. Vascul. Pharmacol. 2019, 112, 24–30. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, D.; Li, S.; Xu, L.; Zhao, J.; Liu, H.; Lu, T.; Zhang, Q. Identification and analysis of differentially expressed microRNAs in rainbow trout (Oncorhynchus mykiss) responding to infectious hematopoietic necrosis virus infection. Dev. Comp. Immunol. 2018, 88, 28–36. [Google Scholar] [CrossRef]
- Ordas, A.; Kanwal, Z.; Lindenberg, V.; Rougeot, J.; Mink, M.; Spaink, H.P.; Meijer, A.H. MicroRNA-146 function in the innate immune transcriptome response of zebrafish embryos to Salmonella typhimurium infection. BMC Genom. 2013, 14, 696. [Google Scholar] [CrossRef] [PubMed]
- Bjørgen, H.; Koppang, E.O. Anatomy of teleost fish immune structures and organs. Immunogenetics 2021, 73, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Fu, J.; Yao, Y.; Xu, M. Identification and characterization of immune-related microRNAs in hybrid snakehead(Channa maculata♀ × Channa argus♂)after treated by Echinacea purpurea (Linn.) Moench. Fish Shellfish. Immunol. 2023, 135, 108653. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, Y.; Wen, L.; Liu, Z.; Yan, F.; Gao, C. Identification and characterization of MicroRNAs in the spleen of common carp immune organ. J. Cell. Biochem. 2014, 115, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Liu, X.; Luo, M.; Yang, L.; Zhu, Z.; Weng, S.; He, J.; Xu, X. miR-10c Facilitates White Spot Syndrome Virus Infection by Targeting Toll3 in Litopenaeus vannemei. Front. Immunol. 2021, 12, 733730. [Google Scholar] [CrossRef] [PubMed]
- Tahamtan, A.; Teymoori-Rad, M.; Nakstad, B.; Salimi, V. Anti-Inflammatory MicroRNAs and Their Potential for Inflammatory Diseases Treatment. Front. Immunol. 2018, 9, 1377. [Google Scholar] [CrossRef]
- Rasal, K.D.; Iquebal, M.A.; Pandey, A.; Behera, P.; Jaiswal, S.; Vasam, M.; Dixit, S.; Raza, M.; Sahoo, L.; Nandi, S.; et al. Revealing liver specific microRNAs linked with carbohydrate metabolism of farmed carp, Labeo rohita (Hamilton, 1822). Genomics 2020, 112, 32–44. [Google Scholar] [CrossRef]
- Mennigen, J.A.; Martyniuk, C.J.; Seiliez, I.; Panserat, S.; Skiba-Cassy, S. Metabolic consequences of microRNA-122 inhibition in rainbow trout, Oncorhynchus mykiss. BMC Genom. 2014, 15, 70. [Google Scholar] [CrossRef]
- Huang, C.; Chen, N.; Wu, X.; Huang, C.; He, Y.; Tang, R.; Wang, W.; Wang, H. The zebrafish miR-462/miR-731 cluster is induced under hypoxic stress via hypoxia-inducible factor 1α and functions in cellular adaptations. FASEB J. 2015, 29, 4901–4913. [Google Scholar] [CrossRef]
- Schyth, B.D.; Bela-Ong, D.B.; Jalali, S.A.H.; Kristensen, L.B.J.; Einer-Jensen, K.; Pedersen, F.S.; Lorenzen, N. Two Virus-Induced MicroRNAs Known Only from Teleost Fishes Are Orthologues of MicroRNAs Involved in Cell Cycle Control in Humans. PLoS ONE 2015, 10, e0132434. [Google Scholar] [CrossRef]
- Schäfer, N.; Matoušek, J.; Rebl, A.; Stejskal, V.; Brunner, R.M.; Goldammer, T.; Verleih, M.; Korytář, T. Effects of Chronic Hypoxia on the Immune Status of Pikeperch (Sander lucioperca Linnaeus, 1758). Biology 2021, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Medley, J.C.; Panzade, G.; Zinovyeva, A.Y. microRNA strand selection: Unwinding the rules. Wiley Interdiscip. Rev. RNA 2021, 12, e1627. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Group | miRs (Total) |
|---|---|
| Eyed eggs | 374 |
| Larvae 2 dph | 375 |
| Larvae 15 dph | 372 |
| Larvae 26 dph | 371 |
| Spleen Fingerling 3 g | 363 |
| Spleen Fingerling 35 g | 350 |
| Head Kidney | 368 |
| Spleen | 366 |
| Liver | 325 |
| Gills | 357 |
| miRNA | Fold Change | Adj. p-Value | |
|---|---|---|---|
| Eyed eggs and larvae 2 dph vs. larvae 15 dph and 26 dph | |||
| Enriched in eyed eggs and larvae 2 dph: | |||
| slu-mir-430b-2-3p | 268.78 | 7.66 × 10−26 | |
| slu-mir-430d-1-2-3p | 235.77 | 1.43 × 10−26 | |
| slu-mir-430bc-3-3p | 222.98 | 9.14 × 10−27 | |
| slu-mir-430bc-2-3p | 181.61 | 4.02 × 10−24 | |
| slu-mir-430c-1-3-5p | 73.14 | 4.10 × 10−18 | |
| slu-mir-430b-1-4-5p | 64.95 | 9.95 × 10−15 | |
| slu-mir-430a-5p | 59.58 | 2.55 × 10−15 | |
| slu-mir-nov2ab-5p | 57.85 | 1.68 × 10−15 | |
| slu-mir-nov2ab-3p | 21.87 | 1.85 × 10−15 | |
| slu-mir-219a-2-3p | 20.55 | 5.57 × 10−18 | |
| Enriched in larvae 15 and 26 dph: | |||
| slu-let-7d-1-5p | 20.04 | 9.67 × 10−17 | |
| slu-let-7f-1-5p | 17.99 | 1.02 × 10−14 | |
| slu-let-7h-1-5p | 17.38 | 4.83 × 10−14 | |
| slu-mir-726-3p | 15.09 | 4.52 × 10−23 | |
| slu-mir-194a-5p | 12.49 | 1.59 × 10−23 | |
| slu-mir-31-5p | 12.05 | 1.40 × 10−16 | |
| slu-mir-726-5p | 11.41 | 1.37 × 10−14 | |
| slu-mir-146-5p | 9.40 | 1.21 × 10−8 | |
| slu-mir-194b-5p | 9.33 | 1.67 × 10−13 | |
| slu-let-7b-1-2-5p | 8.59 | 3.52 × 10−10 | |
| Spleen fingerlings 3 g versus Spleen fingerlings 35 g | |||
| Enriched in fingerling 3 g | |||
| slu-mir-nov6-3p | 50.40 | 1.60 × 10−3 | |
| slu-mir-183-1-2-5p | 46.21 | 2.02 × 10−5 | |
| slu-mir-8159-5p | 20.85 | 3.15 × 10−3 | |
| slu-mir-216c-5p | 19.59 | 1.91 × 10−2 | |
| slu-mir-216b-5p | 19.50 | 1.91 × 10−2 | |
| slu-mir-216a-1-2-5p | 15.50 | 2.50 × 10−2 | |
| slu-mir-375-3p | 15.35 | 8.39 × 10−4 | |
| slu-mir-122-5p | 14.25 | 3.36 × 10−5 | |
| slu-mir-200b-3p | 5.11 | 3.25 × 10−2 | |
| slu-mir-182-5p | 4.79 | 3.29 × 10−2 | |
| Enriched in fingerling 35 g | |||
| slu-mir-1388-3p | 6.68 | 2.45 × 10−4 | |
| slu-mir-730-5p | 5.32 | 5.11 × 10−4 | |
| slu-mir-7132-5p | 5.15 | 5.11 × 10−4 | |
| slu-mir-731-5p | 3.23 | 4.14 × 10−2 | |
| slu-mir-125-1-3-5p | 3.22 | 1.92 × 10−2 | |
| slu-mir-99b-5p | 3.13 | 2.50 × 10−2 | |
| Head kidney versus all other adult tissues | |||
| Enriched in head kidney | |||
| slu-mir-10b-5p | 165.58 | 1.77 × 10−9 | |
| slu-mir-551-3p | 55.04 | 3.44 × 10−3 | |
| slu-mir-10c-5p | 6.90 | 3.89 × 10−2 | |
| Spleen versus all other adult tissues | |||
| Enriched in spleen | |||
| slu-mir-202-5p | 24.59 | 2.03 × 10−2 | |
| slu-mir-187-3p | 20.70 | 3.52 × 10−2 | |
| slu-mir-2187-3p | 18.55 | 6.82 × 10−4 | |
| Gills versus all other adult tissues | |||
| Enriched in gills | |||
| slu-mir-205-5p | 585.85 | 3.67 × 10−14 | |
| slu-mir-203b-3p | 579.26 | 1.08 × 10−18 | |
| slu-mir-725-3p | 447.41 | 1.44 × 10−14 | |
| slu-mir-203a-3p | 398.99 | 2.42 × 10−40 | |
| slu-mir-206-3p | 144.42 | 2.29 × 10−27 | |
| slu-mir-205-3p | 115.31 | 1.45 × 10−15 | |
| slu-mir-184ab-3p | 73.51 | 3.44 × 10−11 | |
| slu-mir-190b-5p | 42.67 | 2.76 × 10−6 | |
| slu-mir-375-3p | 32.95 | 2.86 × 10−2 | |
| slu-mir-182-5p | 25.44 | 2.51 × 10−17 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verleih, M.; Visnovska, T.; Nguinkal, J.A.; Rebl, A.; Goldammer, T.; Andreassen, R. The Discovery and Characterization of Conserved and Novel miRNAs in the Different Developmental Stages and Organs of Pikeperch (Sander lucioperca). Int. J. Mol. Sci. 2024, 25, 189. https://doi.org/10.3390/ijms25010189
Verleih M, Visnovska T, Nguinkal JA, Rebl A, Goldammer T, Andreassen R. The Discovery and Characterization of Conserved and Novel miRNAs in the Different Developmental Stages and Organs of Pikeperch (Sander lucioperca). International Journal of Molecular Sciences. 2024; 25(1):189. https://doi.org/10.3390/ijms25010189
Chicago/Turabian StyleVerleih, Marieke, Tina Visnovska, Julien A. Nguinkal, Alexander Rebl, Tom Goldammer, and Rune Andreassen. 2024. "The Discovery and Characterization of Conserved and Novel miRNAs in the Different Developmental Stages and Organs of Pikeperch (Sander lucioperca)" International Journal of Molecular Sciences 25, no. 1: 189. https://doi.org/10.3390/ijms25010189
APA StyleVerleih, M., Visnovska, T., Nguinkal, J. A., Rebl, A., Goldammer, T., & Andreassen, R. (2024). The Discovery and Characterization of Conserved and Novel miRNAs in the Different Developmental Stages and Organs of Pikeperch (Sander lucioperca). International Journal of Molecular Sciences, 25(1), 189. https://doi.org/10.3390/ijms25010189