
The Role of Sirtuin 6 in the Deacetylation of Histone Proteins as a Factor in the Progression of Neoplastic Disease


Abstract
:1. Introduction
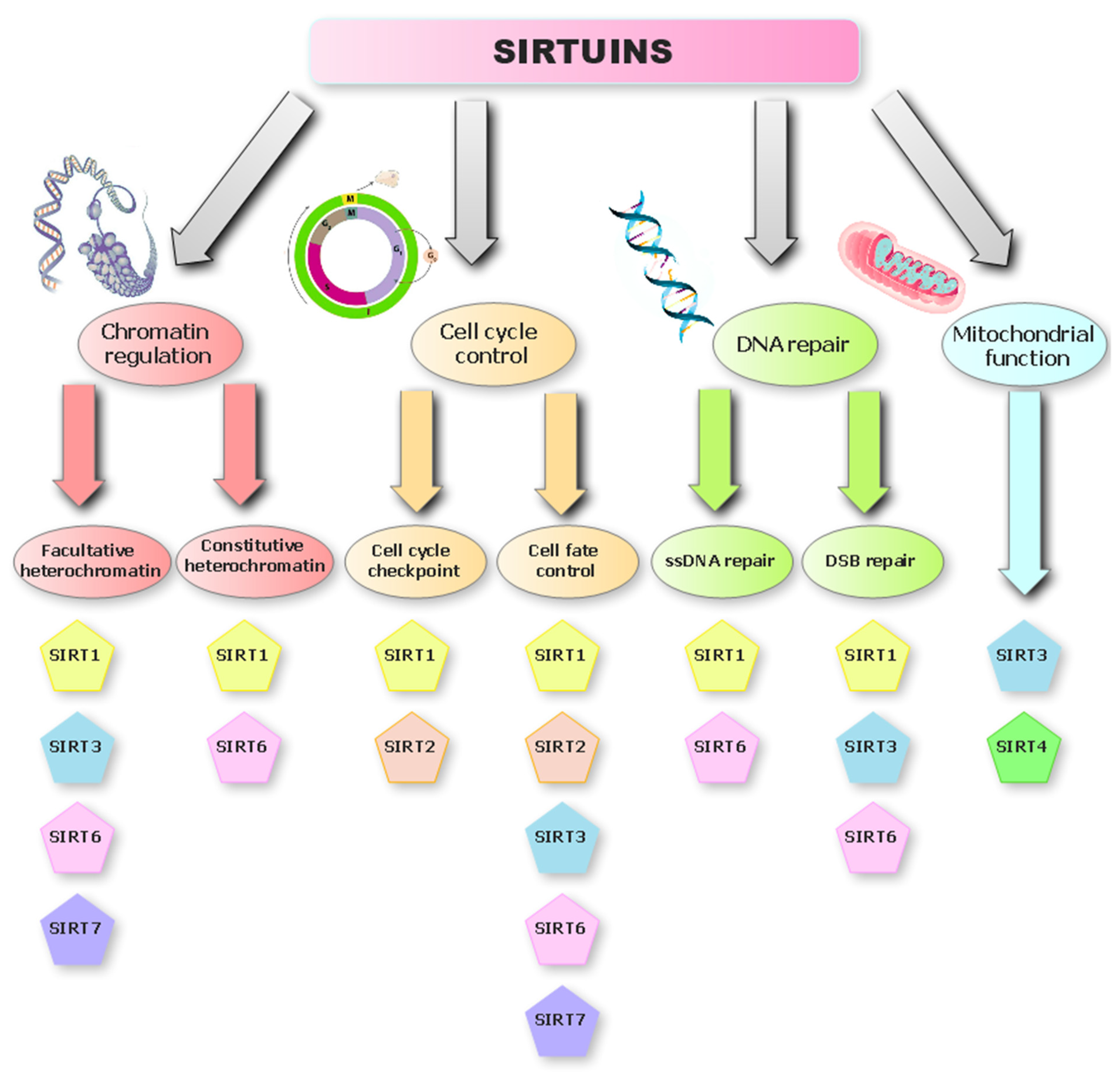
Overview of the Sirtuin Deacetylases
2. SIRT6 Structure
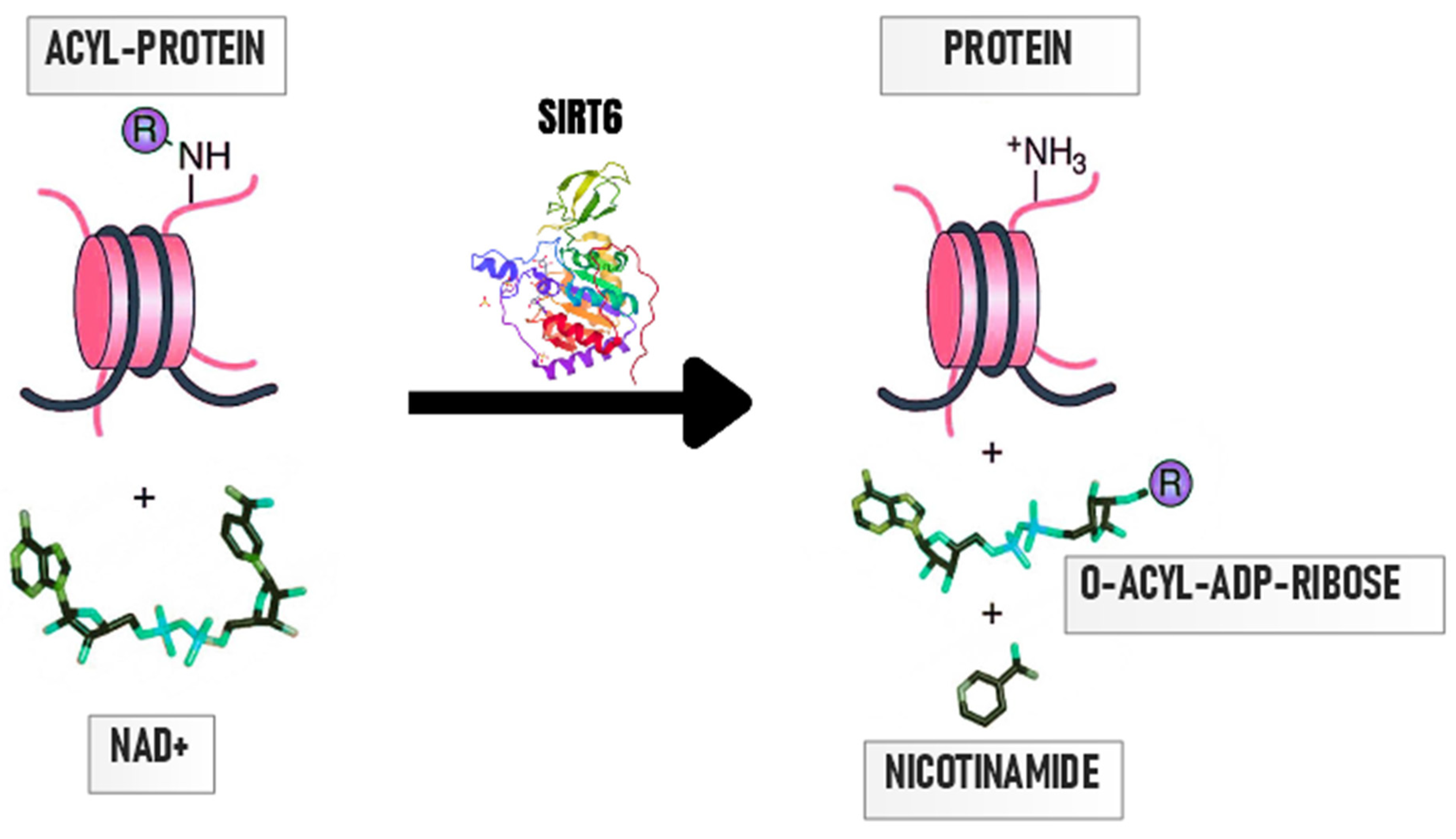
3. Importance of Protein Deacetylation by SIRT6
3.1. The Role of SIRT6-Mediated Protein Deacetylation in Carcinogenesis
3.1.1. The Role of SIRT6 as an Oncogene
3.1.2. The Role of SIRT6 as a Tumor Suppressor
4. Screening of SIRT6 Inhibitors and Activators
4.1. SIRT6 Activators
4.1.1. 4H-Chromen
4.1.2. MDL-811
4.1.3. MDL-800
4.1.4. Fluvastatin
4.2. SIRT6 Inhibitors
4.2.1. Quercetin
4.2.2. Quinazolinedione Derivatives
5. Summary of Sirtuins as a Potential Therapeutic Target
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of Non-Histone Proteins Modulates Cellular Signalling at Multiple Levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Guo, P.; Chen, W.; Li, H.; Li, M.; Li, L. The Histone Acetylation Modifications of Breast Cancer and Their Therapeutic Implications. Pathol. Oncol. Res. 2018, 24, 807–813. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone Deacetylase 6 in Cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Demyanenko, S.; Sharifulina, S. The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses. Int. J. Mol. Sci. 2021, 22, 7947. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of Cellular Processes by Histone and Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Wu, Q.-J.; Zhang, T.-N.; Chen, H.-H.; Yu, X.-F.; Lv, J.-L.; Liu, Y.-Y.; Liu, Y.-S.; Zheng, G.; Zhao, J.-Q.; Wei, Y.-F.; et al. The Sirtuin Family in Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Togni, L.; Mascitti, M.; Sartini, D.; Campagna, R.; Pozzi, V.; Salvolini, E.; Offidani, A.; Santarelli, A.; Emanuelli, M. Nicotinamide N-Methyltransferase in Head and Neck Tumors: A Comprehensive Review. Biomolecules 2021, 11, 1594. [Google Scholar] [CrossRef]
- Campagna, R.; Vignini, A. NAD+ Homeostasis and NAD+-Consuming Enzymes: Implications for Vascular Health. Antioxidants 2023, 12, 376. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Spinelli, G.; Sartini, D.; Milanese, G.; Galosi, A.B.; Emanuelli, M. The Utility of Nicotinamide N-Methyltransferase as a Potential Biomarker to Predict the Oncological Outcomes for Urological Cancers: An Update. Biomolecules 2021, 11, 1214. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Ortega, Á.; Lozano Terol, G.; Sola Martínez, R.A.; Cánovas Díaz, M.; de Diego Puente, T. Bacterial Sirtuins Overview: An Open Niche to Explore. Front. Microbiol. 2021, 12, 744416. [Google Scholar] [CrossRef]
- Palmirotta, R.; Cives, M.; Della-Morte, D.; Capuani, B.; Lauro, D.; Guadagni, F.; Silvestris, F. Sirtuins and Cancer: Role in the Epithelial-Mesenchymal Transition. Oxid. Med. Cell. Longev. 2016, 2016, 3031459. [Google Scholar] [CrossRef]
- Zhao, E.; Hou, J.; Ke, X.; Abbas, M.N.; Kausar, S.; Zhang, L.; Cui, H. The Roles of Sirtuin Family Proteins in Cancer Progression. Cancers 2019, 11, 1949. [Google Scholar] [CrossRef]
- Klein, M.A.; Denu, J.M. Biological and Catalytic Functions of Sirtuin 6 as Targets for Small-Molecule Modulators. J. Biol. Chem. 2020, 295, 11021–11041. [Google Scholar] [CrossRef]
- Costa-Machado, L.F.; Fernandez-Marcos, P.J. The Sirtuin Family in Cancer. Cell Cycle 2019, 18, 2164–2196. [Google Scholar] [CrossRef]
- Kupis, W.; Pałyga, J.; Tomal, E.; Niewiadomska, E. The Role of Sirtuins in Cellular Homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin Functions and Modulation: From Chemistry to the Clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar] [CrossRef]
- Li, Y.; Jin, J.; Wang, Y. SIRT6 Widely Regulates Aging, Immunity, and Cancer. Front. Oncol. 2022, 12, 861334. [Google Scholar] [CrossRef]
- Pan, P.W.; Feldman, J.L.; Devries, M.K.; Dong, A.; Edwards, A.M.; Denu, J.M. Structure and Biochemical Functions of SIRT6. J. Biol. Chem. 2011, 286, 14575–14587. [Google Scholar] [CrossRef]
- You, W.; Steegborn, C. Structural Basis for Activation of Human Sirtuin 6 by Fluvastatin. ACS Med. Chem. Lett. 2020, 11, 2285–2289. [Google Scholar] [CrossRef]
- Fiorentino, F.; Carafa, V.; Favale, G.; Altucci, L.; Mai, A.; Rotili, D. The Two-Faced Role of SIRT6 in Cancer. Cancers 2021, 13, 1156. [Google Scholar] [CrossRef]
- Fan, Y.; Cheng, J.; Yang, Q.; Feng, J.; Hu, J.; Ren, Z.; Yang, H.; Yang, D.; Ding, G. Sirt6-Mediated Nrf2/HO-1 Activation Alleviates Angiotensin II-Induced DNA DSBs and Apoptosis in Podocytes. Food Funct. 2021, 12, 7867–7882. [Google Scholar] [CrossRef]
- Van Meter, M.; Mao, Z.; Gorbunova, V.; Seluanov, A. SIRT6 Overexpression Induces Massive Apoptosis in Cancer Cells but Not in Normal Cells. Cell Cycle 2011, 10, 3153–3158. [Google Scholar] [CrossRef]
- Zhang, Z.-G.; Qin, C.-Y. Sirt6 Suppresses Hepatocellular Carcinoma Cell Growth via Inhibiting the Extracellular Signal-regulated Kinase Signaling Pathway. Mol. Med. Rep. 2014, 9, 882–888. [Google Scholar] [CrossRef]
- Ran, L.-K.; Chen, Y.; Zhang, Z.-Z.; Tao, N.-N.; Ren, J.-H.; Zhou, L.; Tang, H.; Chen, X.; Chen, K.; Li, W.-Y.; et al. SIRT6 Overexpression Potentiates Apoptosis Evasion in Hepatocellular Carcinoma via BCL2-Associated X Protein–Dependent Apoptotic Pathway. Clin. Cancer Res. 2016, 22, 3372–3382. [Google Scholar] [CrossRef]
- Chircop, M.; Speidel, D. Molecular Mechanisms of Cellular Stress Responses in Cancer and Their Therapeutic Implications; Frontiers Media SA: Lausanne, Switzerland, 2015; ISBN 9782889194964. [Google Scholar]
- Chang, M.; Qiao, L.; Li, B.; Wang, J.; Zhang, G.; Shi, W.; Liu, Z.; Gu, N.; Di, Z.; Wang, X.; et al. Suppression of SIRT6 by miR-33a Facilitates Tumor Growth of Glioma through Apoptosis and Oxidative Stress Resistance. Oncol. Rep. 2017, 38, 1251–1258. [Google Scholar] [CrossRef]
- Ouyang, L.; Yi, L.; Li, J.; Yi, S.; Li, S.; Liu, P.; Yang, X. SIRT6 Overexpression Induces Apoptosis of Nasopharyngeal Carcinoma by Inhibiting NF-κB Signaling. Onco. Targets. Ther. 2018, 11, 7613–7624. [Google Scholar] [CrossRef]
- Desantis, V.; Lamanuzzi, A.; Vacca, A. The Role of SIRT6 in Tumors. Haematologica 2018, 103, 1–4. [Google Scholar] [CrossRef]
- Bae, J.S.; Noh, S.J.; Kim, K.M.; Park, S.-H.; Hussein, U.K.; Park, H.S.; Park, B.-H.; Ha, S.H.; Lee, H.; Chung, M.J.; et al. SIRT6 Is Involved in the Progression of Ovarian Carcinomas via β-Catenin-Mediated Epithelial to Mesenchymal Transition. Front. Oncol. 2018, 8, 538. [Google Scholar] [CrossRef]
- Lefort, K.; Brooks, Y.; Ostano, P.; Cario-André, M.; Calpini, V.; Guinea-Viniegra, J.; Albinger-Hegyi, A.; Hoetzenecker, W.; Kolfschoten, I.; Wagner, E.F.; et al. A miR-34a-SIRT6 Axis in the Squamous Cell Differentiation Network. EMBO J. 2013, 32, 2248–2263. [Google Scholar] [CrossRef]
- Ming, M.; Han, W.; Zhao, B.; Sundaresan, N.R.; Deng, C.-X.; Gupta, M.P.; He, Y.-Y. SIRT6 Promotes COX-2 Expression and Acts as an Oncogene in Skin Cancer. Cancer Res. 2014, 74, 5925–5933. [Google Scholar] [CrossRef]
- Jiao, J.; Mikulec, C.; Ishikawa, T.-O.; Magyar, C.; Dumlao, D.S.; Dennis, E.A.; Fischer, S.M.; Herschman, H. Cell-Type-Specific Roles for COX-2 in UVB-Induced Skin Cancer. Carcinogenesis 2014, 35, 1310–1319. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Adamia, S.; Acharya, C.; Tai, Y.-T.; Fulciniti, M.; Ohguchi, H.; Munshi, A.; Acharya, P.; Bhasin, M.K.; et al. Evidence for a Role of the Histone Deacetylase SIRT6 in DNA Damage Response of Multiple Myeloma Cells. Blood 2016, 127, 1138–1150. [Google Scholar] [CrossRef]
- Sebastián, C.; Zwaans, B.M.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor That Controls Cancer Metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef]
- Lin, Z.; Yang, H.; Tan, C.; Li, J.; Liu, Z.; Quan, Q.; Kong, S.; Ye, J.; Gao, B.; Fang, D. USP10 Antagonizes c-Myc Transcriptional Activation through SIRT6 Stabilization to Suppress Tumor Formation. Cell Rep. 2013, 5, 1639–1649. [Google Scholar] [CrossRef]
- Min, L.; Ji, Y.; Bakiri, L.; Qiu, Z.; Cen, J.; Chen, X.; Chen, L.; Scheuch, H.; Zheng, H.; Qin, L.; et al. Liver Cancer Initiation Is Controlled by AP-1 through SIRT6-Dependent Inhibition of Survivin. Nat. Cell Biol. 2012, 14, 1203–1211. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Das, S. SIRT6 Deacetylates PKM2 to Suppress Its Nuclear Localization and Oncogenic Functions. Proc. Natl. Acad. Sci. USA 2016, 113, E538–E547. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Fischer, K.; Baus, K.; Kashyap, A.; Ma, S.; Krupp, M.; Linke, M.; Teufel, A.; Zechner, U.; Strand, D.; et al. Sirtuin-6-Dependent Genetic and Epigenetic Alterations Are Associated with Poor Clinical Outcome in Hepatocellular Carcinoma Patients. Hepatology 2013, 58, 1054–1064. [Google Scholar] [CrossRef]
- Zhang, J.; Yin, X.-J.; Xu, C.-J.; Ning, Y.-X.; Chen, M.; Zhang, H.; Chen, S.-F.; Yao, L.-Q. The Histone Deacetylase SIRT6 Inhibits Ovarian Cancer Cell Proliferation via down-Regulation of Notch 3 Expression. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 818–824. [Google Scholar]
- Choe, M.; Brusgard, J.L.; Chumsri, S.; Bhandary, L.; Zhao, X.F.; Lu, S.; Goloubeva, O.G.; Polster, B.M.; Fiskum, G.M.; Girnun, G.D.; et al. The RUNX2 Transcription Factor Negatively Regulates SIRT6 Expression to Alter Glucose Metabolism in Breast Cancer Cells. J. Cell. Biochem. 2015, 116, 2210–2226. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional Silencing and Longevity Protein Sir2 Is an NAD-Dependent Histone Deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Rahnasto-Rilla, M.; Tyni, J.; Huovinen, M.; Jarho, E.; Kulikowicz, T.; Ravichandran, S.; Bohr, V.A.; Ferrucci, L.; Lahtela-Kakkonen, M.; Moaddel, R. Natural Polyphenols as Sirtuin 6 Modulators. Sci. Rep. 2018, 8, 4163. [Google Scholar] [CrossRef]
- Tenhunen, J.; Kučera, T.; Huovinen, M.; Küblbeck, J.; Bisenieks, E.; Vigante, B.; Ogle, Z.; Duburs, G.; Doležal, M.; Moaddel, R.; et al. Screening of SIRT6 Inhibitors and Activators: A Novel Activator Has an Impact on Breast Cancer Cells. Biomed. Pharmacother. 2021, 138, 111452. [Google Scholar] [CrossRef]
- He, T.; Shang, J.; Gao, C.; Guan, X.; Chen, Y.; Zhu, L.; Zhang, L.; Zhang, C.; Zhang, J.; Pang, T. A Novel SIRT6 Activator Ameliorates Neuroinflammation and Ischemic Brain Injury via EZH2/FOXC1 Axis. Acta Pharm. Sin. B 2021, 11, 708–726. [Google Scholar] [CrossRef]
- Shang, J.; Zhu, Z.; Chen, Y.; Song, J.; Huang, Y.; Song, K.; Zhong, J.; Xu, X.; Wei, J.; Wang, C.; et al. Small-Molecule Activating SIRT6 Elicits Therapeutic Effects and Synergistically Promotes Anti-Tumor Activity of Vitamin D3 in Colorectal Cancer. Theranostics 2020, 10, 5845–5864. [Google Scholar] [CrossRef]
- Fiorentino, F.; Mai, A.; Rotili, D. Emerging Therapeutic Potential of SIRT6 Modulators. J. Med. Chem. 2021, 64, 9732–9758. [Google Scholar] [CrossRef]
- Huang, Z.; Zhao, J.; Deng, W.; Chen, Y.; Shang, J.; Song, K.; Zhang, L.; Wang, C.; Lu, S.; Yang, X.; et al. Identification of a Cellularly Active SIRT6 Allosteric Activator. Nat. Chem. Biol. 2018, 14, 1118–1126. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Liu, Q.; Huang, Y.; Li, R.; Wu, T.; Zhang, Z.; Zhou, J.; Huang, H.; Tang, Q.; et al. Sirt6 Alleviated Liver Fibrosis by Deacetylating Conserved Lysine 54 on Smad2 in Hepatic Stellate Cells. Hepatology 2021, 73, 1140–1157. [Google Scholar] [CrossRef]
- Cai, Y.; Zhao, F. Fluvastatin Suppresses the Proliferation, Invasion, and Migration and Promotes the Apoptosis of Endometrial Cancer Cells by Upregulating Sirtuin 6 (SIRT6). Bioengineered 2021, 12, 12509–12520. [Google Scholar] [CrossRef]
- Liu, T.; Li, Z.; Tian, F. Quercetin Inhibited the Proliferation and Invasion of Hepatoblastoma Cells through Facilitating SIRT6-Medicated FZD4 Silence. Hum. Exp. Toxicol. 2021, 40, S96–S107. [Google Scholar] [CrossRef]
- You, W.; Zheng, W.; Weiss, S.; Chua, K.F.; Steegborn, C. Structural Basis for the Activation and Inhibition of Sirtuin 6 by Quercetin and Its Derivatives. Sci. Rep. 2019, 9, 19176. [Google Scholar] [CrossRef]
- Sociali, G.; Galeno, L.; Parenti, M.D.; Grozio, A.; Bauer, I.; Passalacqua, M.; Boero, S.; Donadini, A.; Millo, E.; Bellotti, M.; et al. Quinazolinedione SIRT6 Inhibitors Sensitize Cancer Cells to Chemotherapeutics. Eur. J. Med. Chem. 2015, 102, 530–539. [Google Scholar] [CrossRef]
- Islam, S.; Abiko, Y.; Uehara, O.; Chiba, I. Sirtuin 1 and Oral Cancer. Oncol. Lett. 2019, 17, 729–738. [Google Scholar]
- Alhazzazi, T.Y.; Kamarajan, P.; Xu, Y.; Ai, T.; Chen, L.; Verdin, E.; Kapila, Y.L. A Novel Sirtuin-3 Inhibitor, LC-0296, Inhibits Cell Survival and Proliferation, and Promotes Apoptosis of Head and Neck Cancer Cells. Anticancer Res. 2016, 36, 49–60. [Google Scholar]
- Ezhilarasan, D.; Lakshmi, T.; Subha, M.; Deepak Nallasamy, V.; Raghunandhakumar, S. The Ambiguous Role of Sirtuins in Head and Neck Squamous Cell Carcinoma. Oral Dis. 2022, 28, 559–567. [Google Scholar] [CrossRef]
- Choi, H.-K.; Cho, K.B.; Phuong, N.T.T.; Han, C.Y.; Han, H.-K.; Hien, T.T.; Choi, H.S.; Kang, K.W. SIRT1-Mediated FoxO1 Deacetylation Is Essential for Multidrug Resistance-Associated Protein 2 Expression in Tamoxifen-Resistant Breast Cancer Cells. Mol. Pharm. 2013, 10, 2517–2527. [Google Scholar] [CrossRef]
- Khongkow, M.; Olmos, Y.; Gong, C.; Gomes, A.R.; Monteiro, L.J.; Yagüe, E.; Cavaco, T.B.; Khongkow, P.; Man, E.P.S.; Laohasinnarong, S.; et al. SIRT6 Modulates Paclitaxel and Epirubicin Resistance and Survival in Breast Cancer. Carcinogenesis 2013, 34, 1476–1486. [Google Scholar] [CrossRef]
- Sinha, S.; Sharma, S.; Vora, J.; Shrivastava, N. Emerging Role of Sirtuins in Breast Cancer Metastasis and Multidrug Resistance: Implication for Novel Therapeutic Strategies Targeting Sirtuins. Pharmacol. Res. 2020, 158, 104880. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Cancer | Level of SIRT6 Expression |
|---|---|
| Ovarian Cancer (OC) | ↑ |
| Skin Cancer (SCC) | ↑ |
| Hepatocellular Carcinoma (HCC) | ↓ |
| Breast Cancer (BC) | ↓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baran, M.; Miziak, P.; Stepulak, A.; Cybulski, M. The Role of Sirtuin 6 in the Deacetylation of Histone Proteins as a Factor in the Progression of Neoplastic Disease. Int. J. Mol. Sci. 2024, 25, 497. https://doi.org/10.3390/ijms25010497
Baran M, Miziak P, Stepulak A, Cybulski M. The Role of Sirtuin 6 in the Deacetylation of Histone Proteins as a Factor in the Progression of Neoplastic Disease. International Journal of Molecular Sciences. 2024; 25(1):497. https://doi.org/10.3390/ijms25010497
Chicago/Turabian StyleBaran, Marzena, Paulina Miziak, Andrzej Stepulak, and Marek Cybulski. 2024. "The Role of Sirtuin 6 in the Deacetylation of Histone Proteins as a Factor in the Progression of Neoplastic Disease" International Journal of Molecular Sciences 25, no. 1: 497. https://doi.org/10.3390/ijms25010497
APA StyleBaran, M., Miziak, P., Stepulak, A., & Cybulski, M. (2024). The Role of Sirtuin 6 in the Deacetylation of Histone Proteins as a Factor in the Progression of Neoplastic Disease. International Journal of Molecular Sciences, 25(1), 497. https://doi.org/10.3390/ijms25010497