
Comparative In Vitro Dissolution Assessment of Calcined and Uncalcined Hydroxyapatite Using Differences in Bioresorbability and Biomineralization

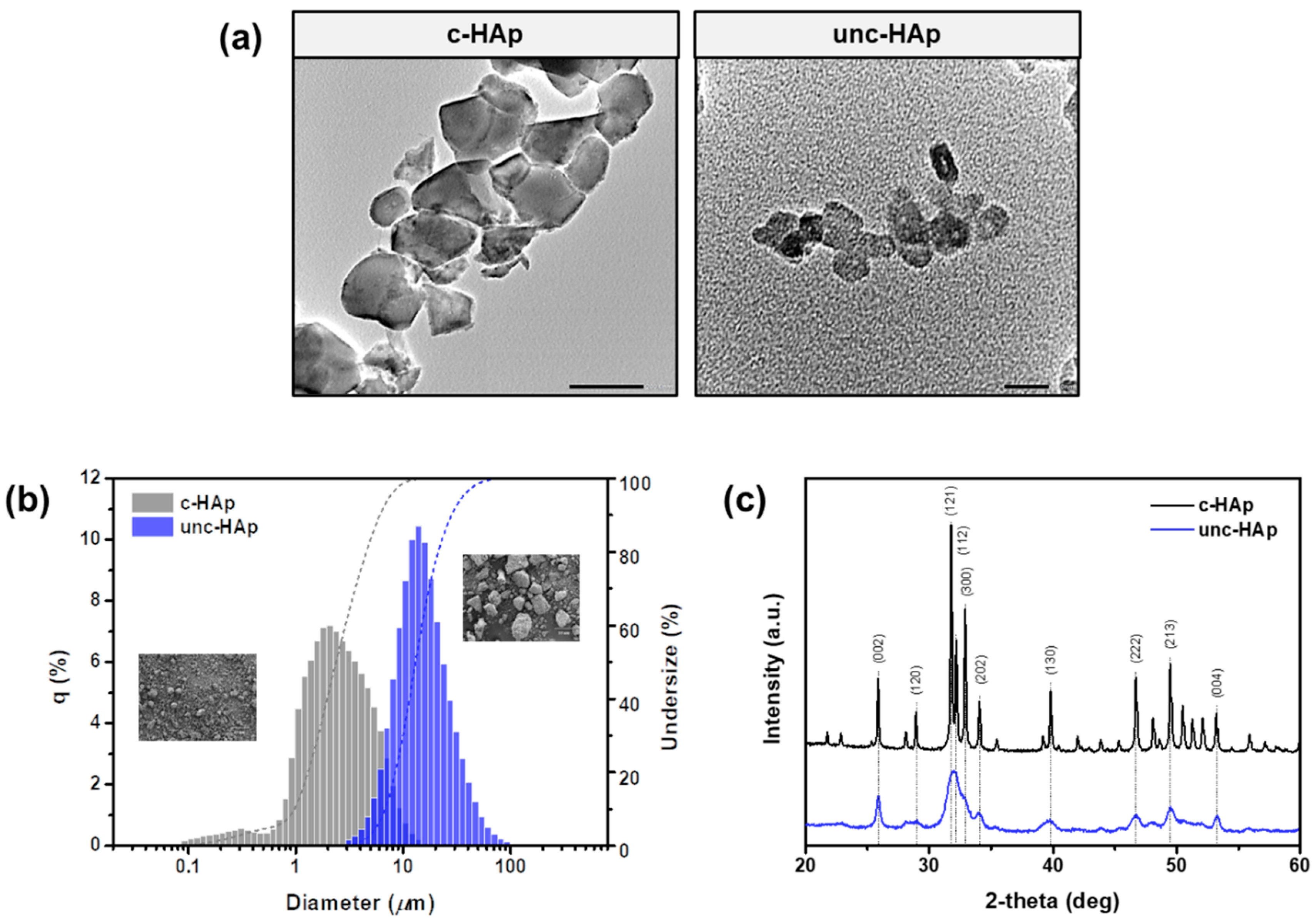{kind=link}
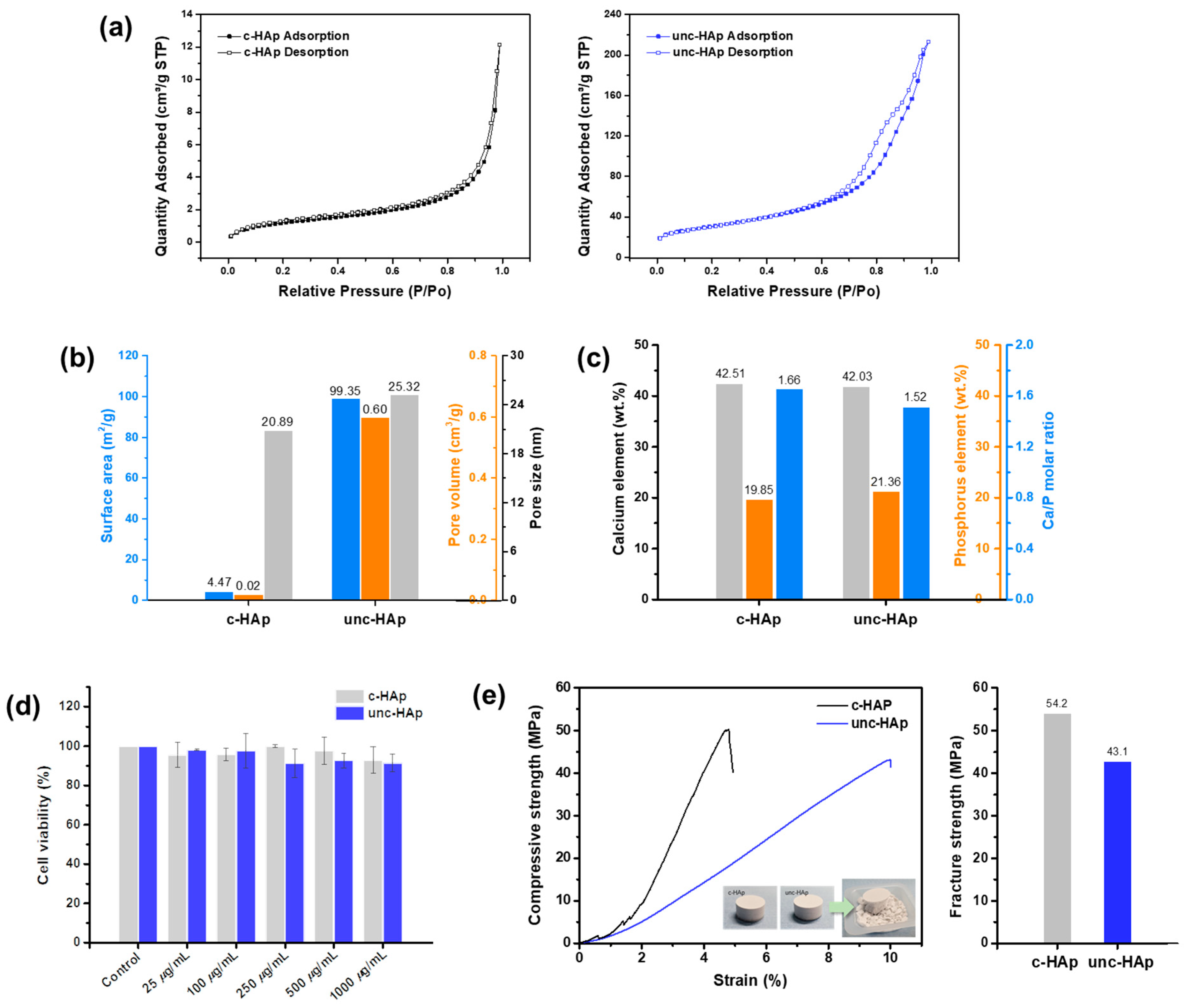{kind=link}
{kind=link}
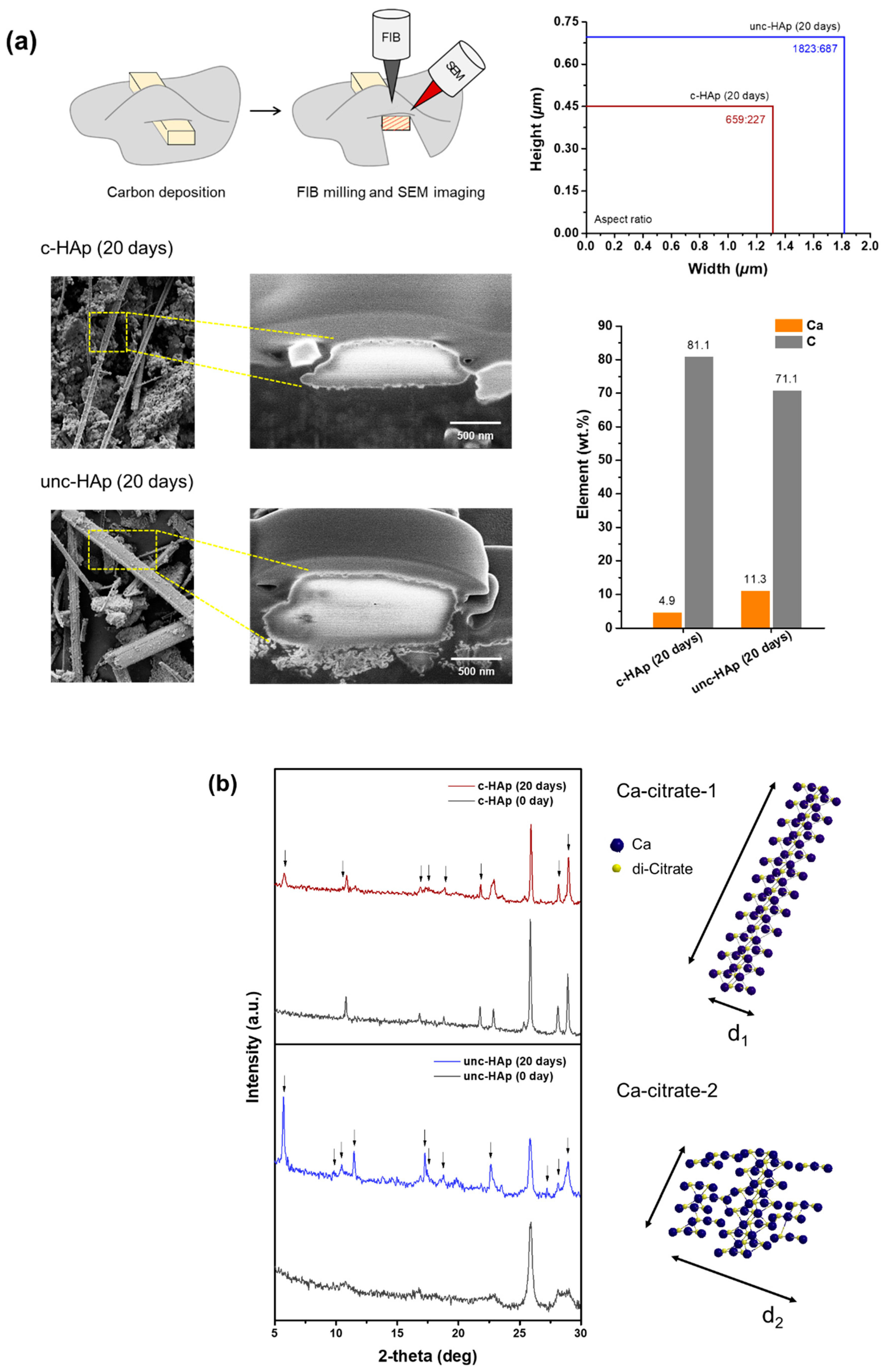{kind=link}
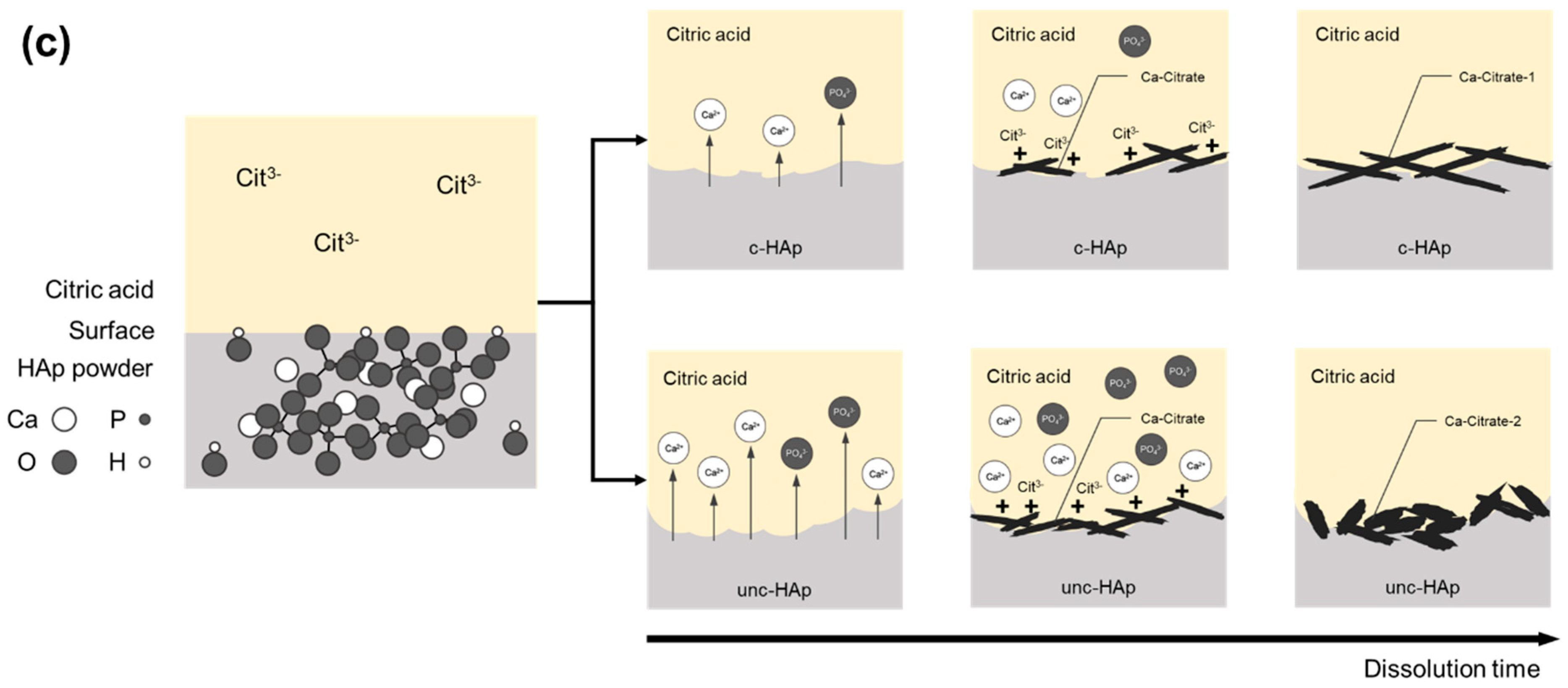{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
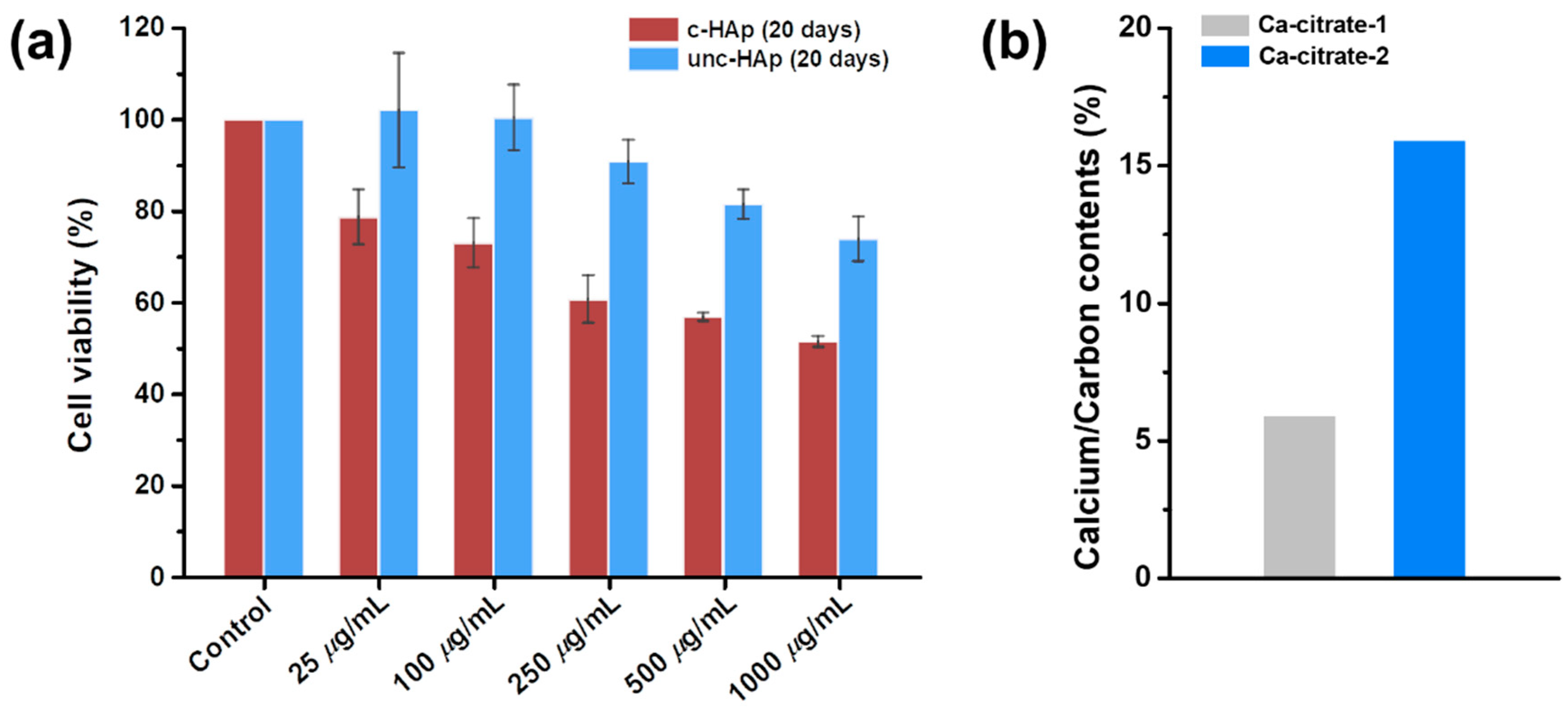
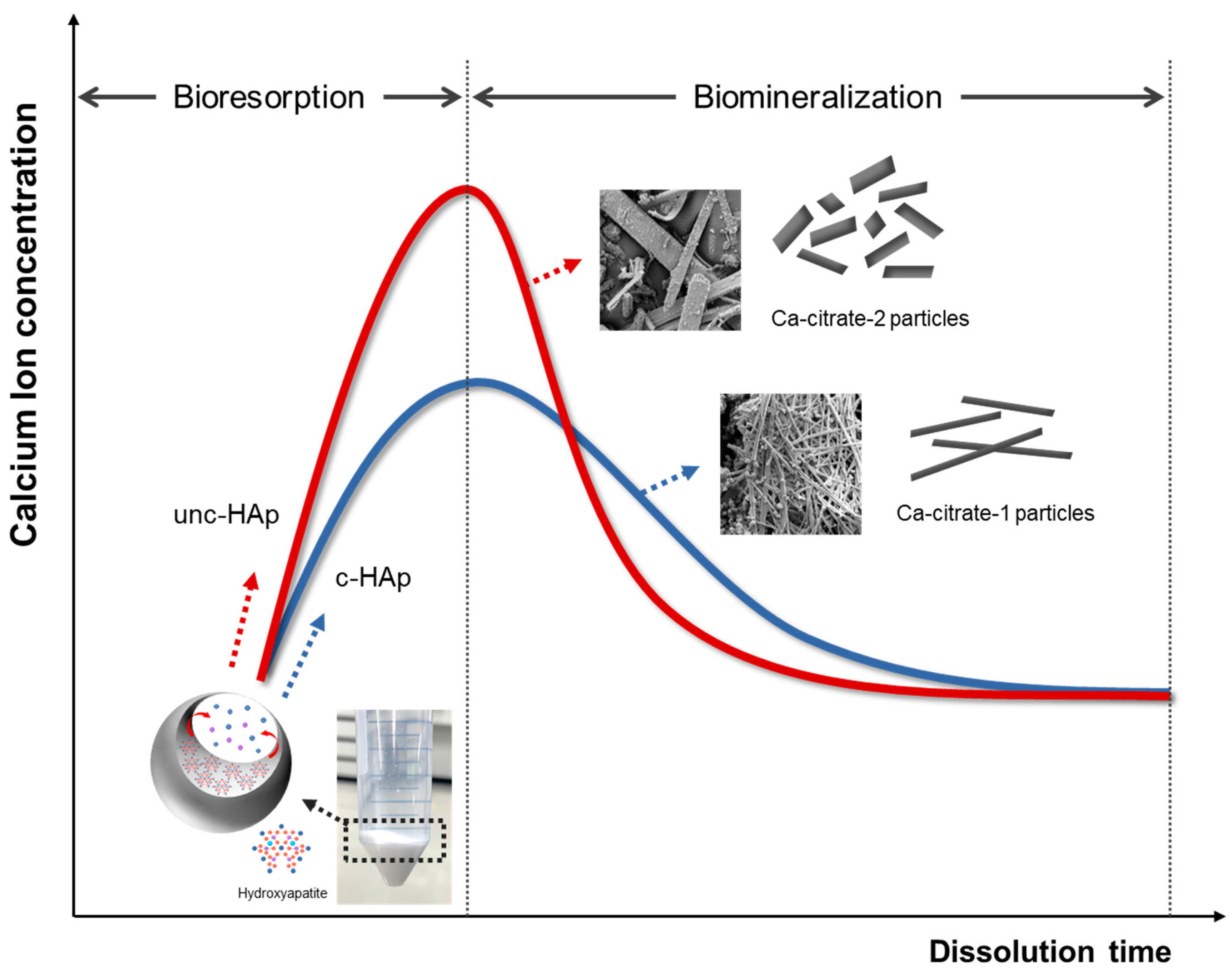
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Preparation of Uncalcined Hydroxyapatite (Unc-HAp) and Calcined Hydroxyapatite (c-HAp)
3.3. Cell Viability
3.4. In Vitro Dissolution Assessments
3.5. Characterizations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, X.; Kolzow, J.; Chen, R.R.; Du, J. Effect of solution condition on hydroxyapatite formation in evaluating bioactivity of B2O3 containing 45S5 bioactive glasses. Bioact. Mater. 2019, 4, 207–214. [Google Scholar] [CrossRef]
- Alorku, K.; Manoj, M.; Yuan, A. A plant-mediated synthesis of nanostructured hydroxyapatite for biomedical applications: A review. RSC Adv. 2020, 10, 40923–40939. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, S.; Agarwal, A.K.; Rai, K.N.; Garg, A. Development of high strength hydroxyapatite by solid-state-sintering process. Ceram. Int. 2007, 33, 419–426. [Google Scholar] [CrossRef]
- Koonawoot, R.; Saelee, C.; Thiansem, S.; Punyanitya, S. Synthesis control and characterization of hydroxyapatite ceramic using a solid state reaction. In Proceedings of the 1st Mae Fah Luang University International Conference, Chiang Rai, Thailand, 29 November–1 December 2012. [Google Scholar]
- Cai, S.; Wang, Y.; Lv, H.; Peng, Z.; Yao, K. Synthesis of carbonated hydroxyapatite nanofibers by mechanochemical methods. Ceram. Int. 2005, 31, 135–138. [Google Scholar] [CrossRef]
- Afshar, A.; Ghorbani, M.; Ehsani, N.; Saeri, M.R.; Sorrell, C.C. Some important factors in the wet precipitation process of hydroxyapatite. Mater. Des. 2003, 24, 197–202. [Google Scholar] [CrossRef]
- Peng, H.; Wang, J.; Lv, S.; Wen, J.; Chen, J.-F. Synthesis and characterization of hydroxyapatite nanoparticles prepared by a high-gravity precipitation method. Ceram. Int. 2015, 41, 14340–14349. [Google Scholar] [CrossRef]
- Nagata, F.; Yamauchi, Y.; Tomita, M.; Kato, K. Hydrothermal synthesis of hydroxyapatite nanoparticles and their protein adsorption behavior. J. Cera. Soc. Jpn. 2013, 121, 797–801. [Google Scholar] [CrossRef]
- Canillas, M.; Rivero, R.; García-Carrodeguas, R.; Barba, F.; Rodríguez, M.A. Processing of hydroxyapatite obtained by combustion synthesis. Bol. Soc. Esp. Ceram. Vidr. 2017, 56, 237–242. [Google Scholar] [CrossRef]
- Cacciotti, I. Cationic and Anionic Substitutions in Hydroxyapatite; Springer International Publishing: Berlin/Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Nakazato, T.; Tsukui, S.; Nakagawa, N.; Kai, T. Continuous production of hydroxyapatite powder by drip pyrolysis in a fluidized bed. Adv. Powder Technol. 2012, 23, 632–639. [Google Scholar] [CrossRef]
- Kottegoda, N.; Sandaruwan, C.; Priyadarshana, G.; Siriwardhana, A.; Rathnayake, U.A.; Berugoda Arachchige, D.M.; Kumarasinghe, A.R.; Dahanayake, D.; Karunaratne, V.; Amaratunga, G.A.J. Urea-Hydroxyapatite Nanohybrids for Slow Release of Nitrogen. ACS Nano 2017, 11, 1214–1221. [Google Scholar] [CrossRef]
- Yang, H.; Zeng, H.; Hao, L.; Zhao, N.; Du, C.; Liao, H.; Wang, Y. Effects of hydroxyapatite microparticle morphology on bone mesenchymal stem cell behavior. J. Mater. Chem. B 2014, 2, 4703–4710. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, O.; Ipekoglu, M.; Mahmutyzicioglu, N.; Varmis, M.; Kaya, E.; Altintas, S. Preparation and characterization of porous hydroxyapatite pellets: Effects of calcination and sintering on the porous structure and mechanical properties. Proc. Inst. Mech. Eng. Part L J. Mater. Des. Appl. 2015, 230, 985–993. [Google Scholar] [CrossRef]
- Paul, W.; Sharma, C.P. Development of porous spherical hydroxyapatite granules: Application towards protein delivery. J. Mater. Sci. Mater. Med. 1999, 10, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.H.; Son, J.S.; Kim, K.M.; Han, M.; Oh, D.S.; Lee, Y.K. Drug-loaded porous spherical hydroxyapatite granules for bone regeneration. J. Mater. Sci. Mater. Med. 2011, 22, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Hernández, A.K.; Martínez-Juárez, J.; Gervacio-Arciniega, J.J.; Silva-González, R.; Robles-Águila, M.J. Effect of Ultrasound Irradiation on the Synthesis of Hydroxyapatite/Titanium Oxide Nanocomposites. Crystals 2020, 10, 959. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Z.; Joseph, J.; Zhang, X.; Ferdows, B.E.; Patel, D.N.; Chen, W.; Banfi, G.; Molinaro, R.; Cosco, D.; et al. Biomaterials and nanomedicine for bone regeneration: Progress and future prospects. Exploration 2021, 1, 20210011. [Google Scholar] [CrossRef] [PubMed]
- Girón, J.; Kerstner, E.; Medeiros, T.; Oliveira, L.; Machado, G.M.; Malfatti, C.F.; Pranke, P. Biomaterials for bone regeneration: An orthopedic and dentistry overview. Braz. J. Med. Biol. Res. 2021, 54, e11055. [Google Scholar] [CrossRef]
- Tang, G.; Liu, Z.; Liu, Y.; Yu, J.; Wang, X.; Tan, Z.; Ye, X. Recent Trends in the Development of Bone Regenerative Biomaterials. Front. Cell Dev. Biol. 2021, 9, 665813. [Google Scholar] [CrossRef]
- Le Gars Santoni, B.; Niggli, L.; Dolder, S.; Loeffel, O.; Sblendorio, G.A.; Heuberger, R.; Maazouz, Y.; Stähli, C.; Döbelin, N.; Bowen, P.; et al. Effect of minor amounts of β-calcium pyrophosphate and hydroxyapatite on the physico-chemical properties and osteoclastic resorption of β-tricalcium phosphate cylinders. Bioact. Mater. 2022, 10, 222–235. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, L.; Liu, Z.; Su, J.; Tang, Y.; Tan, P.; Zhu, X.; Zhang, K.; Ma, X.; Jiang, J.; et al. Canine ACL reconstruction with an injectable hydroxyapatite/collagen paste for accelerated healing of tendon-bone interface. Bioact. Mater. 2023, 20, 1–15. [Google Scholar] [CrossRef]
- Li, J.; Deng, C.; Liang, W.; Kang, F.; Bai, Y.; Ma, B.; Wu, C.; Dong, S. Mn-containing bioceramics inhibit osteoclastogenesis and promote osteoporotic bone regeneration via scavenging ROS. Bioact. Mater. 2021, 6, 3839–3850. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Kanno, T.; Tatsumi, H.; Miyamoto, K.; Sha, J.; Hideshima, K.; Matsuzaki, Y. Feasibility of a Three-Dimensional Porous Uncalcined and Unsintered Hydroxyapatite/poly-d/l-lactide Composite as a Regenerative Biomaterial in Maxillofacial Surgery. Materials 2018, 11, 2047. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.N.; Kanno, T.; Bai, Y.; Sha, J.; Hideshima, K. Bone Regeneration Potential of Uncalcined and Unsintered Hydroxyapatite/Poly l-lactide Bioactive/Osteoconductive Sheet Used for Maxillofacial Reconstructive Surgery: An In Vivo Study. Materials 2019, 12, 2931. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Tatsumi, H.; Karino, M.; Yoshino, A.; Koike, T.; Ide, T.; Sekine, J. Applicability of an Unsintered Hydroxyapatite Particles/Poly-L-Lactide Composite Sheet with Tack Fixation for Orbital Fracture Reconstruction. J. Hard Tissue Biol. 2016, 25, 329–334. [Google Scholar] [CrossRef]
- Morizane, K.; Shikinami, Y.; Fujibayashi, S.; Goto, K.; Otsuki, B.; Kawai, T.; Shimizu, T.; Matsuda, S. Implantable composite devices of unsintered hydroxyapatite and poly-l-lactide with dispersive marbling morphology to enhance in vivo bioactivity and bioresorbability. Mater. Sci. Eng. C 2019, 97, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, S.; Dong, Q.N.; Ngo, H.X.; Bai, Y.; Sha, J.; Toda, E.; Okui, T.; Kanno, T. Bioactive Regeneration Potential of the Newly Developed Uncalcined/Unsintered Hydroxyapatite and Poly-l-Lactide-Co-Glycolide Biomaterial in Maxillofacial Reconstructive Surgery: An In Vivo Preliminary Study. Materials 2021, 14, 2461. [Google Scholar] [CrossRef] [PubMed]
- Shikinami, Y.; Okuno, M. Bioresorbable devices made of forged composites of hydroxyapatite (HA) particles and poly-L-lactide (PLLA): Part I. Basic characteristics. Biomaterials 1999, 20, 859–877. [Google Scholar] [CrossRef]
- Shikinami, Y.; Okuno, M. Bioresorbable devices made of forged composites of hydroxyapatite (HA) particles and poly L-lactide (PLLA). Part II: Practical properties of miniscrews and miniplates. Biomaterials 2002, 22, 3197–3211. [Google Scholar] [CrossRef]
- Shikinami, Y.; Matsusue, Y.; Nakamura, T. The complete process of bioresorption and bone replacement using devices made of forged composites of raw hydroxyapatite particles/poly l-lactide (F-u-HA/PLLA). Biomaterials 2005, 26, 5542–5551. [Google Scholar] [CrossRef]
- Corina, G.; Janis, L.; Matteo, D.E.; Gerard, D.; Aurora, M.; Cecilia, R.; Ossi, H.; Tomoaia, C.M. Advanced Mg, Zn, Sr, Si Multi-Substituted Hydroxyapatites for Bone Regeneration. Int. J. Nanomed. 2020, 15, 1037–1058. [Google Scholar] [CrossRef]
- Bauer, L.; Rogina, A.; Ivankovi’c, M.; Ivankovi’c, H. Medical-Grade Poly(Lactic Acid)/Hydroxyapatite Composite Films: Thermal and In Vitro Degradation Properties. Polymers 2023, 15, 1512. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.X.; Dong, Q.N.; Bai, Y.; Sha, J.; Ishizuka, S.; Okui, T.; Sukegawa, S.; Kanno, T. Bone Regeneration Capacity of Newly Developed Uncalcined/Unsintered Hydroxyapatite and Poly-L-lactide-co-glycolide Sheet in Maxillofacial Surgery: An In Vivo Study. Nano Mater. 2021, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Todo, M.; Ito, H. Mechanical Properties and Degradability of HA/PLLA Composites with Different Particle Size Distribution. In Interface Oral Health Science 2011; Sasaki, K., Suzuki, O., Takahashi, N., Eds.; Springer: Tokyo, Japan, 2012; pp. 100–101. [Google Scholar]
- Sukegawa, S.; Kanno, T.; Katase, N.; Shibata, A.; Takahashi, Y.; Furuki, Y. Clinical Evaluation of an Unsintered Hydroxyapatite/Poly-L-Lactide Osteoconductive Composite Device for the Internal Fixation of Maxillofacial Fractures. J. Craniofacial Surg. 2016, 27, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Lee, J.H. Clinical courses and degradation patterns of absorbable plates in facial bone fracture patients. Arch. Craniofacial Surg. 2019, 20, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lee, J.K.; Moursi, A.; Lannutti, J.J. Ca/P ratio effects on the degradation of hydroxyapatite in vitro. J. Biomed. Mater. Res. A 2003, 67, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xiong, J.; Liu, J.; Zhu, W.; Wang, D. Investigation of the In Vitro Degradation of a Novel Polylactide/Nanohydroxyapatite Composite for Artificial Bone. J. Nano Mater. 2013, 2013, 515741. [Google Scholar] [CrossRef]
- Jiang, L.; Xiong, C.; Jiang, L.; Xu, L. Degradation behavior of hydroxyapatite/poly(lactic-co-glycolic) acid nanocomposite in simulated body fluid. Mater. Res. Bull. 2013, 48, 4186–4190. [Google Scholar] [CrossRef]
- Kaygili, O.; Keser, S.; Kom, M.; Bulut, N.; Dorozhkin, S.V. The effect of simulating body fluid on the structural properties of hydroxyapatite synthesized in the presence of citric acid. Prog. Biomater. 2016, 5, 173–182. [Google Scholar] [CrossRef]
- You, B.C.; Meng, C.E.; Mohd Nasir, N.F.; Mohd Tarmizi, E.Z.; Fhan, K.S.; Kheng, E.S.; Abdul Majid, M.S.; Mohd Jamir, M.R. Dielectric and biodegradation properties of biodegradable nano-hydroxyapatite/starch bone scaffold. J. Mater. Res. Technol. 2022, 18, 3215–3226. [Google Scholar] [CrossRef]
- Antoniac, I.V.; Antoniac, A.; Vasile, E.; Tecu, C.; Fosca, M.; Yankova, V.G.; Rau, J.V. In vitro characterization of novel nanostructured collagen-hydroxyapatite composite scaffolds doped with magnesium with improved biodegradation rate for hard tissue regeneration. Bioact. Mater. 2021, 6, 3383–3395. [Google Scholar] [CrossRef]
- Zhang, Y.; Shu, T.; Wang, S.; Liu, Z.; Cheng, Y.; Li, A.; Pei, D. The Osteoinductivity of Calcium Phosphate-Based Biomaterials: A Tight Interaction With Bone Healing. Front. Bioeng. Biotechnol. 2022, 10, 911180. [Google Scholar] [CrossRef] [PubMed]
- Alt, V.; Cheung, W.H.; Chow, S.K.; Thormann, U.; Cheung, E.N.; Lips, K.S.; Schnettler, R.; Leung, K.S. Bone formation and degradation behavior of nanocrystalline hydroxyapatite with or without collagen-type 1 in osteoporotic bone defects-an experimental study in osteoporotic goats. Injury 2016, 47 (Suppl. S2), S58–S65. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Gonzalo Juan, I.; Arango-Ospina, M.; Riedel, R.; Boccaccini, A.R.; Ionescu, E. Apatite Forming Ability and Dissolution Behavior of Boron- and Calcium-Modified Silicon Oxycarbides in Comparison to Silicate Bioactive Glass. ACS Biomater. Sci. Eng. 2019, 5, 5337–5347. [Google Scholar] [CrossRef] [PubMed]
- Rezwan, K.; Chen, Q.Z.; Blaker, J.J.; Boccaccini, A.R. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, J.O.; Battistone, G.C. Biodegradable bone repair materials. Synthetic polymers and ceramics. Clin. Orthop. Relat. Res. 1986, 207, 290–305. [Google Scholar] [CrossRef]
- ISO 10993-14; Biological Evaluation of Medical Devices-Part 14: Identification and Quantification of Degradation Products from Ceramics. International Organization for Standardization: Geneva, Switzerland, 2001.
- Mohd Pu’ad, N.A.S.; Abdul Haq, R.H.; Mohd Noh, H.; Abdullah, H.Z.; Idris, M.I.; Lee, T.C. Synthesis method of hydroxyapatite: A review. Mater. Today Proc. 2020, 29, 233–239. [Google Scholar] [CrossRef]
- Ma, G. Three common preparation methods of hydroxyapatite. IOP Conf. Ser. Mater. Sci. Eng. 2019, 688, 033057. [Google Scholar] [CrossRef]
- Ramirez-Gutierrez, C.F.; Londoño-Restrepo, S.M.; del Real, A.; Mondragón, M.A.; Rodriguez-García, M.E. Effect of the temperature and sintering time on the thermal, structural, morphological, and vibrational properties of hydroxyapatite derived from pig bone. Ceram. Int. 2017, 43, 7552–7559. [Google Scholar] [CrossRef]
- Guo, L.; Huang, M.; Zhang, X. Effects of sintering temperature on structure of hydroxyapatite studied with Rietveld method. J. Mater. Sci. Mater. Med. 2003, 14, 817–822. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Yuan, Y.; Cai, F.; Yang, L. Pore structure characteristics and fractal structure evaluation of medium- and high-rank coal. Energy Explor. Exploit. 2022, 40, 328–342. [Google Scholar] [CrossRef]
- Mohd Pu’ad, N.A.S.; Koshy, P.; Abdullah, H.Z.; Idris, M.I.; Lee, T.C. Syntheses of hydroxyapatite from natural sources. Heliyon 2019, 5, e01588. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Singh, R.; Teh, Y.C.; Tan, Y.M.; Yap, B.K. The Effects of Calcium-to-Phosphorus Ratio on the Densification and Mechanical Properties of Hydroxyapatite Ceramic. Int. J. Appl. Ceram. Technol. 2015, 12, 223–227. [Google Scholar] [CrossRef]
- Liu, H.; Yazici, H.; Ergun, C.; Webster, T.J.; Bermek, H. An in vitro evaluation of the Ca/P ratio for the cytocompatibility of nano-to-micron particulate calcium phosphates for bone regeneration. Acta Biomater. 2008, 4, 1472–1479. [Google Scholar] [CrossRef]
- Pellegrino, E.D.; Biltz, R.M. Bone Carbonate and the Ca to P Molar Ratio. Nature 1968, 219, 1261–1262. [Google Scholar] [CrossRef] [PubMed]
- de Groot, K. Bioceramics of Calcium Phosphate; CRC Press: New York, NY, USA, 1983. [Google Scholar]
- Hench, L.L. Bioceramics: From Concept to Clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- Barros, A.; Pires, R.; Mano, J.F.; Reis, R.L. Advances in Calcium Phosphate Biomaterials; Springer: Berlin/Heidelberg, Germany, 2014; Volume 2. [Google Scholar] [CrossRef]
- Doumeng, M.; Makhlouf, L.; Berthet, F.; Marsan, O.; Delbé, K.; Denape, J.; Chabert, F. A comparative study of the crystallinity of polyetheretherketone by using density, DSC, XRD, and Raman spectroscopy techniques. Polym. Test. 2021, 93, 106878. [Google Scholar] [CrossRef]
- Rotaru, R.; Savin, M.; Tudorachi, N.; Peptu, C.; Samoila, P.; Sacarescu, L.; Harabagiu, V. Ferromagnetic iron oxide–cellulose nanocomposites prepared by ultrasonication. Polym. Chem. 2018, 9, 860–868. [Google Scholar] [CrossRef]
- Bano, N.; Jikan, S.; Basri, H.; Adzila, S.; Dagaci, M. XRD and FTIR study of A&B type carbonated hydroxyapatite extracted from bovine bone. AIP Conf. Proc. 2019, 2068, 020100. [Google Scholar] [CrossRef]
- Castillo-Paz, A.M.; Londoño-Restrepo, S.M.; Tirado-Mejía, L.; Mondragón, M.A.; Rodríguez-García, M.E. Nano to micro size transition of hydroxyapatite in porcine bone during heat treatment with low heating rates. Prog. Nat. Sci. Mater. Int. 2020, 30, 494–501. [Google Scholar] [CrossRef]
- Wang, D.; Xie, Y.; Jaisi, D.P.; Jin, Y. Effects of low-molecular-weight organic acids on the dissolution of hydroxyapatite nanoparticles. Environ. Sci. Nano 2016, 3, 768–779. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Ruiz-Agudo, C.; Di Lorenzo, F.; Alvarez-Lloret, P.; Ibañez-Velasco, A.; Rodriguez-Navarro, C. Citrate Stabilizes Hydroxylapatite Precursors: Implications for Bone Mineralization. ACS Biomater. Sci. Eng. 2021, 7, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Pastero, L.; Bruno, M.; Aquilano, D. Habit Change of Monoclinic Hydroxyapatite Crystals Growing from Aqueous Solution in the Presence of Citrate Ions: The Role of 2D Epitaxy. Crystals 2018, 8, 308. [Google Scholar] [CrossRef]
- Herdtweck, E.; Kornprobst, T.; Sieber, R.; Straver, L.; Plank, J. Crystal Structure, Synthesis, and Properties of tri-Calcium di-Citrate tetra-Hydrate [Ca3(C6H5O7)2(H2O)2]·2H2O. Z. Anorg. Allg. Chem. 2011, 637, 655–659. [Google Scholar] [CrossRef]
- Kaduk, J.A. Crystal structures of tricalcium citrates. Powder Diffr. 2018, 33, 98–107. [Google Scholar] [CrossRef]
- Lee, M.N.; Hwang, H.-S.; Oh, S.-H.; Roshanzadeh, A.; Kim, J.-W.; Song, J.H.; Kim, E.-S.; Koh, J.-T. Elevated extracellular calcium ions promote proliferation and migration of mesenchymal stem cells via increasing osteopontin expression. Exp. Mol. Med. 2018, 50, 1–16. [Google Scholar] [CrossRef]
- Chai, Y.C.; Carlier, A.; Bolander, J.; Roberts, S.J.; Geris, L.; Schrooten, J.; Van Oosterwyck, H.; Luyten, F.P. Current views on calcium phosphate osteogenicity and the translation into effective bone regeneration strategies. Acta Biomater. 2012, 8, 3876–3887. [Google Scholar] [CrossRef]
- Maeno, S.; Niki, Y.; Matsumoto, H.; Morioka, H.; Yatabe, T.; Funayama, A.; Toyama, Y.; Taguchi, T.; Tanaka, J. The effect of calcium ion concentration on osteoblast viability, proliferation and differentiation in monolayer and 3D culture. Biomaterials 2005, 26, 4847–4855. [Google Scholar] [CrossRef]
- Noorani, T.Y.; Luddin, N.; Rahman, I.A.; Masudi, S.M. In Vitro Cytotoxicity Evaluation of Novel Nano-Hydroxyapatite-Silica Incorporated Glass Ionomer Cement. J. Clin. Diagn. Res. 2017, 11, Zc105–Zc109. [Google Scholar] [CrossRef]
- Musa, M.; Kannan, T.P.; Masudi, S.a.M.; Rahman, I.A. Assessment of DNA damage caused by locally produced hydroxyapatite-silica nanocomposite using Comet assay on human lung fibroblast cell line. Mol. Cell. Toxicol. 2012, 8, 53–60. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, W.Y.; Pyun, J.C.; Chang, J.H. Comparative In Vitro Dissolution Assessment of Calcined and Uncalcined Hydroxyapatite Using Differences in Bioresorbability and Biomineralization. Int. J. Mol. Sci. 2024, 25, 621. https://doi.org/10.3390/ijms25010621
Jang WY, Pyun JC, Chang JH. Comparative In Vitro Dissolution Assessment of Calcined and Uncalcined Hydroxyapatite Using Differences in Bioresorbability and Biomineralization. International Journal of Molecular Sciences. 2024; 25(1):621. https://doi.org/10.3390/ijms25010621
Chicago/Turabian StyleJang, Woo Young, Jae Chul Pyun, and Jeong Ho Chang. 2024. "Comparative In Vitro Dissolution Assessment of Calcined and Uncalcined Hydroxyapatite Using Differences in Bioresorbability and Biomineralization" International Journal of Molecular Sciences 25, no. 1: 621. https://doi.org/10.3390/ijms25010621
APA StyleJang, W. Y., Pyun, J. C., & Chang, J. H. (2024). Comparative In Vitro Dissolution Assessment of Calcined and Uncalcined Hydroxyapatite Using Differences in Bioresorbability and Biomineralization. International Journal of Molecular Sciences, 25(1), 621. https://doi.org/10.3390/ijms25010621