5.2.1. Preparation of the Monosaccharide Building Blocks and Block Synthesis
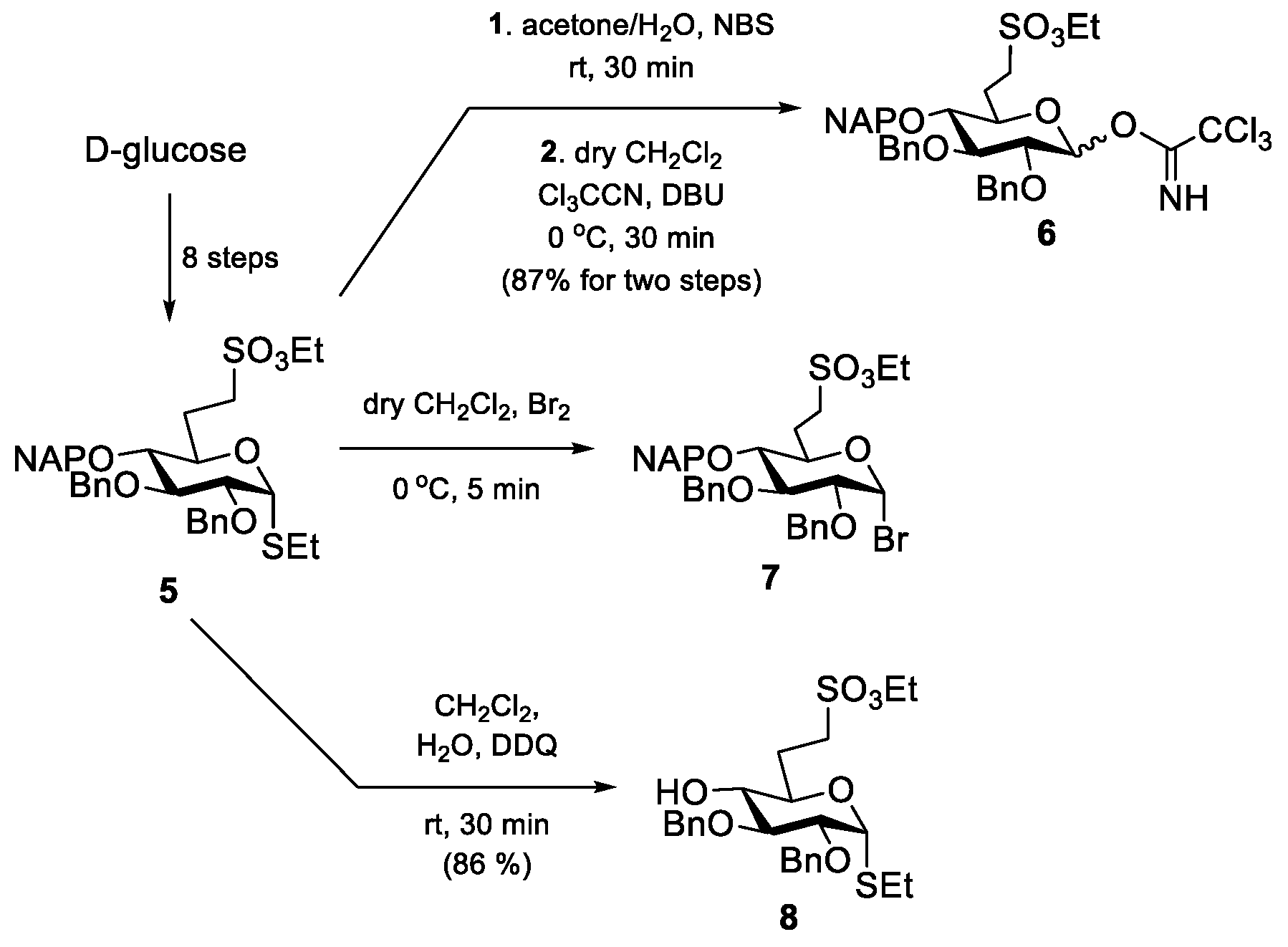
2,3-Di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-4-O-(2-naphthyl)methyl-α,β-
d-glucopyranosyl-trichloroacetimidate (
6). Compound
5 [
24] (102 mg, 0.158 mmol) was converted to
6 according to general method A. The residue was purified by silica gel column chromatography (6:4
n-hexane/EtOAc + 1% Et
3N) to give
6 (89 mg, 87%) as a colourless syrup.
Rf 0.67 (6:4
n-hexane/EtOAc + 1% Et
3N).
Ethyl 2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-1-thio-α-
d-glucopyranoside (
8), Compound
5 [
24] (1.08 g, 1.660 mmol) was converted to
8 according to general method B. The crude product was purified by silica gel column chromatography (6:4
n-hexane/EtOAc) to give
8 (729 mg, 86%) as a colourless syrup.
Rf = 0.50 (
n-hexane/EtOAc 6:4); [α]
D24 = +77.3 (
c = 0.22 in CHCl
3);
1H NMR (400 MHz, CDCl
3):
δ = 7.39–7.27 (m, 10H, Ar-H), 5.32 (d,
J1,2 = 5.4 Hz, 1H, H-1), 4.97 (d,
Jgem = 11.4 Hz, 1H, PhC
H2a), 4.72 (d,
Jgem = 11.6 Hz, 1H, PhC
H2a), 4.65 (d,
Jgem = 11.4 Hz, 1H, PhC
H2b), 4.60 (d,
Jgem = 11.6 Hz, 1H, PhC
H2b), 4.26 (q,
J = 7.1 Hz, 2H, SO
3C
H2CH
3), 4.00 (td,
J = 3.2 Hz,
J = 9.0 Hz, 1H, H-5), 3.75 (dd,
J = 5.4 Hz,
J = 9.5 Hz, 1H, H-2), 3.59 (t,
J2,3=3,4 = 9.1 Hz, 1H, H-3), 3.27–3.19 (m, 2H, H-4, H-7a), 3.09 (ddd,
J = 5.0 Hz,
J = 11.2 Hz,
J = 14.2 Hz, 1H, H-7b), 2.51 (qd,
J = 4.1 Hz,
J = 7.4 Hz, 2H, SC
H2CH
3), 2.40–2.39 (s, 1H, H-4-O
H), 2.38–2.33 (m, 1H, H-6a), 2.01–1.92 (m, 1H, H-6b), 1.37 (t,
J = 7.1 Hz, 3H, SO
3CH
2C
H3), 1.28 (t,
J = 7.4 Hz, 3H, SCH
2C
H3) ppm;
13C NMR (100 MHz, CDCl
3):
δ = 138.5, 137.7 (2C, C
q Ar), 128.7–128.0 (10C, Ar), 83.0 (1C, C-1), 81.4 (1C, C-3), 79.3 (1C, C-2), 75.4 (1C, Ph
CH
2), 73.3 (1C, C-4), 72.1 (1C, Ph
CH
2), 69.1 (1C, C-5), 66.2 (1C, SO
3CH
2CH
3), 46.9 (1C, C-7), 26.1 (1C, C-6), 24.0 (1C, S
CH
2CH
3), 15.2 (1C, SO
3CH
2CH
3), 14.8 (1C, SCH
2CH
3) ppm; UHR ESI-QTOF (positive ion):
m/
z calcd for [C
25H
34O
7S
2 + Na]
+: 533.1638; found: 533.1638.
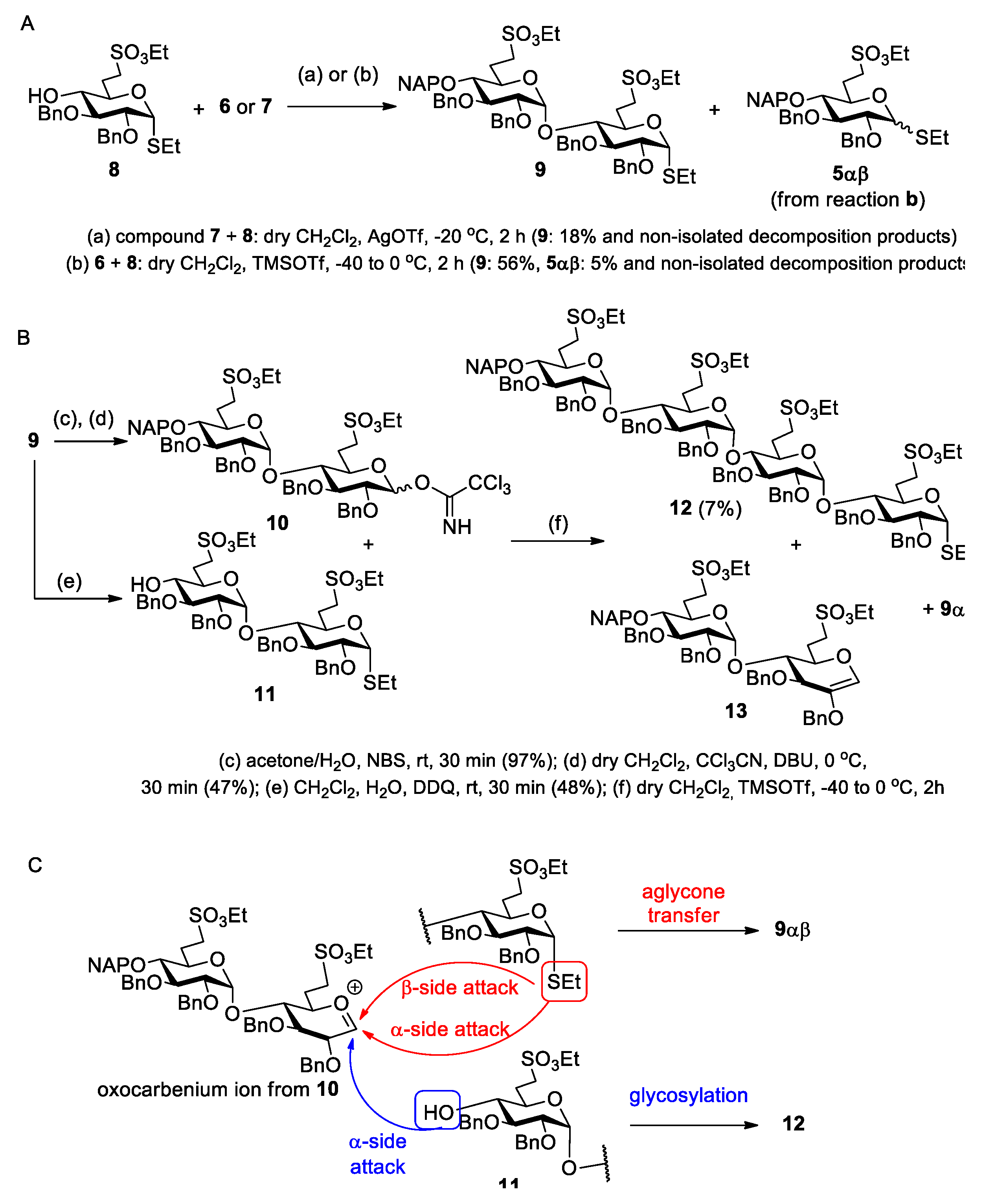
Ethyl [2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-4-O-(2-naphthyl)methyl-α-d-glucopyranosyl]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-1-thio-α-d-glucopyranoside (9).
Method I.: A solution of
5 [
24] (500 mg, 0.769 mmol) in dry CH
2Cl
2 (4.4 mL) was cooled to 0 °C under argon and Br
2 (45 µL) was added. The mixture was stirred for 30 min. The reaction mixture was concentrated at 30 °C and co-evaporated with toluene (2 × 5 mL). The obtained glycosyl bromide (
7) and acceptor
8 (450 mg, 0.880 mmol, 1.15 equiv.) were dissolved in dry CH
2Cl
2 (10 mL) and 4 Å molecular sieves (2.5 g) were added. The solution was stirred for 15 min at room temperature then for a further 15 min at −20 °C. AgOTf (395 mg, 1.538 mmol) dissolved in toluene (2.0 mL) was added and the mixture was allowed to warm up to room temperature in 1.5 h. The reaction mixture was diluted with CH
2Cl
2 (80 mL) and filtered through a pad of Celite. The filtrate was washed successively with satd. aq. solution of NaHCO
3 (2 × 15 mL) and H
2O (2 × 15 mL). The organic phase was dried over MgSO
4 and concentrated. The crude product was purified by column chromatography (7:3
n-hexane/EtOAc) to give
9 (148 mg, 18%) as a colourless syrup.
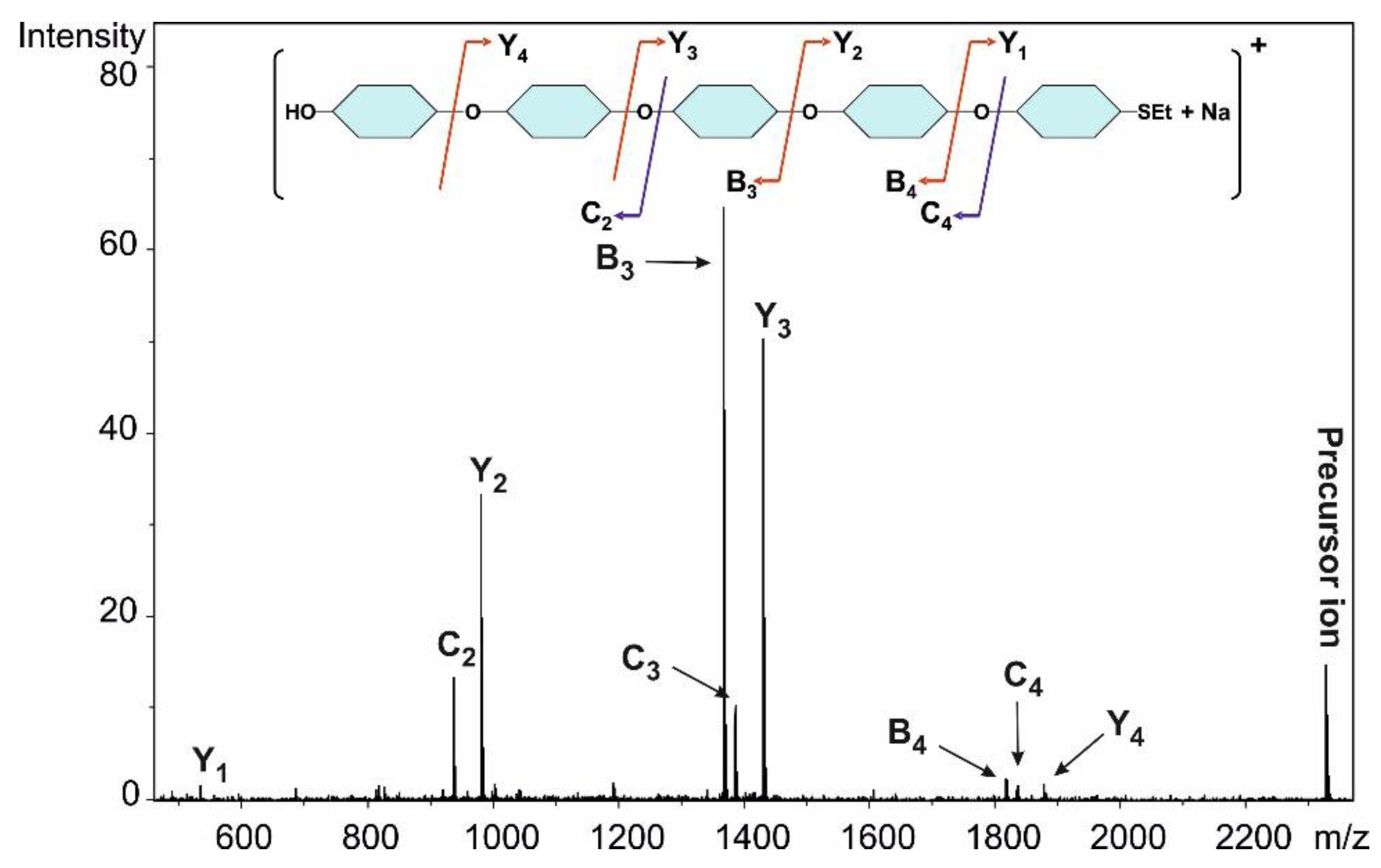
Method II.: To the solution of acceptor 8 (46 mg, 0.090 mmol) and donor 6 (89 mg, 0.137 mmol, 1.5 equiv.) in dry CH2Cl2 (2.9 mL), 4 Å molecular sieves (0.5 g) were added. The stirred mixture was cooled to −40 °C under argon. After 30 min at this temperature, TMSOTf (2.5 µL, 0.013 mmol, 0.1 equiv.) in dry CH2Cl2 (98 µL) was added and the reaction mixture was allowed to warm up to 0 °C in 1.5 h. The mixture was diluted with CH2Cl2 (30 mL) washed successively with satd. aq. solution of NaHCO3 (2 × 5 mL) and H2O (2 × 5 mL), dried over MgSO4 and concentrated. The crude product was purified by column chromatography (7:3 n-hexane/EtOAc) to give 9 (56 mg, 56%) as a colourless syrup and 5αβ (5 mg, 5%) as a colourless syrup. Data of 9: Rf = 0.32 (n-hexane/EtOAc 7:3); [α]D24 = +78.2 (c = 0.11 in CHCl3); 1H NMR (360 MHz, CDCl3): δ = 7.80–7.13 (m, 27H, Ar-H), 5.58 (d, J1′,2′ = 3.8 Hz, 1H, H-1′), 5.27 (d, J1,2 = 5.3 Hz, 1H, H-1), 5.05–4.49 (m, 10H, 4 × PhCH2, NpCH2), 4.20–4.14 (m, 5H, 2 × SO3CH2CH3, H-5), 3.96 (t, J = 9.3 Hz, 1H, H-3′), 3.88 (t, J = 8.9 Hz, 1H, H-3), 3.79 (dd, J = 5.3 Hz, J = 9.1 Hz, 1H, H-2), 3.77 (dd, J = 2.3 Hz, J = 9.3 Hz, 1H, H-5′), 3.50 (dd, J = 8.1 Hz, J = 9.4 Hz, 1H, H-4), 3.47 (dd, J = 3.7 Hz, J = 9.7 Hz, 1H, H-2′), 3.30–3.18 (m, 4H, H-7′a,b, H-7a, H-4′), 3.09–3.00 (m, 1H, H-7b), 2.54–2.45 (m, 3H, SCH2CH3, H-6a), 2.40–2.34 (m, 1H, H-6a’), 1.99–1.89 (m, 2H, H-6b, H-6b’), 1.32–1.18 (m, 9H, 2 × SO3CH2CH3, SCH2CH3) ppm; 13C NMR (90 MHz, CDCl3): δ = 138.6, 138. 8, 138.5, 137.9, 137.5, 135.4, 133.3, 133.2 (7C, Cq Ar), 128.5–126.0 (27C, Ar), 96.9 (C-1′), 82.9 (C-1), 81.6 (1C, C-3), 81.5 (2C, C-3′, C-4′), 79.6 (1C, C-2), 79.5 (1C, C-2′), 77.5 (1C, C-4), 75.7, 75.3, 74.1, 73.7, 72.5 (5C, 4 × PhCH2, NpCH2), 69.8 (1C, C-5′), 68.3 (1C, C-5), 66.7, 66.3 (2C, 2 × SO3CH2CH3), 47.3 (1C, C-7), 46.5 (1C, C-7′), 27.6 (1C, C-6), 26.5 (1C, C-6′), 24.1 (1C, SCH2CH3), 15.2 (2C, 2 × SO3CH2CH3), 14.9 (1C, SCH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C59H70O14S3 + Na]+: 1121.3820; found: 1121.3822.
[2,3-Di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-4-O-(2-naphthyl)methyl-α-d-glucopyranosyl]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α,β-d-glucopyranosyl-trichloroacetimidate (10). Compound 9 (530 mg, 0.482 mmol) was converted to 10 according to general method A. The residue was purified by column chromatography (6:4 n-hexane/EtOAc + 1% Et3N) to give 10 (263 mg, 47%) as a colourless syrup. Rf 0.72 (6:4 n-hexane/EtOAc + 1% Et3N).
Ethyl [2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-1-thio-α-d-glucopyranoside (11) Compound 9 (377 mg, 0.343 mmol) was converted to 11 according to general method B. The crude product was purified by column chromatography (7:3 n-hexane/EtOAc) to give 11 (158 mg, 48%) as a colourless syrup. Rf = 0.22 (n-hexane/EtOAc 7:3); [α]D24 = +79.3 (c = 0.15 in CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.37–7.14 (m, 20H, Ar-H), 5.58 (d, J1′,2′ = 3.6 Hz, 1H, H-1′), 5.28 (d, J1,2 = 5.3 Hz, 1H, H-1), 4.97–4.46 (m, 8H, 4 × PhCH2), 4.27 (q, J = 7.1 Hz, 4H, 2 × SO3CH2CH3), 4.18 (td, J = 2.1 Hz, J = 9.6 Hz, 1H, H-5), 3.88 (t, J = 8.5 Hz, 1H, H-3), 3.81 (dd, J = 5.3 Hz, J = 9.0 Hz, 1H, H-2), 3.73–3.66 (m, 2H, H-3′, H-5′), 3.54 (t, J = 9.1 Hz, 1H, H-4), 3.40 (dd, J = 3.6 Hz, J = 9.7 Hz, 1H, H-2′), 3.33–3.21 (m, 4H, H-4′, H-7a, H-7′a,b), 3.06 (td, J = 4.6 Hz, J = 12.9 Hz, J = 14.4 Hz, 1H, H-7b), 2.52 (q, J = 7.1 Hz, 2H, SCH2CH3), 2.48–2.43 (m, 1H, H-6a), 2.38–2.31 (m, 2H, H-6a’, H-4-OH), 2.03–1.91 (m, 2H, H-6b, H-6b’), 1.39–1.35 (m, 6H, 2 × SO3CH2CH3), 1.30 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (100 MHz, CDCl3): δ = 138.8, 138.4, 137.8, 137.4 (4C, Cq Ar), 128.7–126.6 (20C, Ar), 96.9 (C-1′), 82.9 (C-1), 81.4 (1C, C-3), 81.0 (C-3′), 79.6 (1C, C-2), 79.2 (1C, C-2′), 77.1 (1C, C-4), 75.4, 74.0 (2C, 2 × PhCH2), 73.7 (1C, C-4′), 73.4, 72.5 (2C, 2 × PhCH2),70.3 (1C, C-5′), 68.3 (1C, C-5), 66.7, 66.4 (2C, 2 × SO3CH2CH3), 47.2 (1C, C-7), 46.4 (1C, C-7′), 27.3 (1C, C-6), 26.5 (1C, C-6′), 24.1 (1C, SCH2CH3), 15.2 (2C, 2 × SO3CH2CH3), 14.8 (SCH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C48H62O14S3 + Na]+: 981.3194; found: 981.3198.
Ethyl [2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-4-O-(2′-naphthyl)methyl-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-1-thio-α-d-glucopyranoside (12). To the solution of acceptor 11 (140 mg, 0.146 mmol) and donor 10 (263 mg, 0.219 mmol, 1.5 equiv.) in dry CH2Cl2 (5.0 mL), 4 Å molecular sieves (1.0 g) were added. The stirred mixture was cooled to −40 °C under argon. After 30 min at this temperature, TMSOTf (4.0 µL, 0.022 mmol) in dry CH2Cl2 (100 µL) was added and the mixture was allowed to warm up to 0 °C in 1.5 h. The mixture was diluted with CH2Cl2 (40 mL), washed successively with satd. aq. solution of NaHCO3 (2 × 10 mL) and H2O (2 × 10 mL), dried over MgSO4 and concentrated. The crude product was purified by column chromatography (98:2 CH2Cl2/acetone) to give 12 (21 mg, 7%) as a colourless syrup. Rf = 0.39 (CH2Cl2/acetone 98:2); [α]D24 = +35.0 (c = 0.08 in CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.81–7.02 (m, 47H, Ar-H), 5.47 (d, J = 3.7 Hz, 1H, H-1), 5.42 (d, J = 3.7 Hz, 1H, H-1), 5.28 (d, J = 5.3 Hz, 1H, H-1), 5.23 (d, J = 3.6 Hz, 1H, H-1), 5.04–4.33 (m, 18H, 8 × PhCH2, NpCH2), 4.30–4.14 (m, 9H, H-5, 4 × SO3CH2CH3), 3.98–3.79 (m, 8H, H-2, H-3, H-3′, H-3″, H-3‴, H-5′, H-5″, H-5‴), 3.51–3.39 (m, 6H, H-2′, H-2″, H-2‴, H-4, H-4′, H-4″), 3.34–3.03 (m, 9H, H-4‴, 4 × H-7a,b), 2.56–2.35 (m, 6H, SCH2CH3, 4 × H-6a), 1.96–1.88 (m, 4H, 4 × H-6b), 1.39–1.22 (m, 15H, 4 × SO3CH2CH3, SCH2CH3) ppm; 13C NMR (100 MHz, CDCl3): δ = 138.8, 138.7, 138.6, 138.1, 137.9, 137.8, 137.6, 137.6, 135.5, 133.4, 133.1 (11C, Cq Ar), 128.6–126.0 (47C, Ar), 97.9, 97.8, 97.1 (3C, C-1′, C-1″, C-1‴), 82.9 (1C, C-1), 81.9, 81.5, 81.2, 80.4, 80.3, 80.2, 80.1, 79.5, 79.4, 79.2, 79.1, 78.8, 78.6 (12C, skeleton carbons), 75.6, 75.3, 74.3, 74.2, 73.9, 73.7, 73.6, 72.5 (9C, 8 × PhCH2, NpCH2), 70.1, 69.6, 69.5 (3C, C-5′, C-5″, C-5‴), 68.4 (1C, C-5), 67.2, 67.1, 67.0, 66.5 (4C, 4 × SO3CH2CH3), 47.1, 46.5, 46.4, 46.3 (4C, 4 × C-7), 29.9, 27.4, 26.8, (4C, 4 × C-6), 24.2 (1C, SCH2CH3), 15.4, 15.2, 14.9 (5C, 4 × SO3CH2CH3, SCH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C105H126O28S5 + 2Na]2+: 1020.8428; found: 1020.8439.
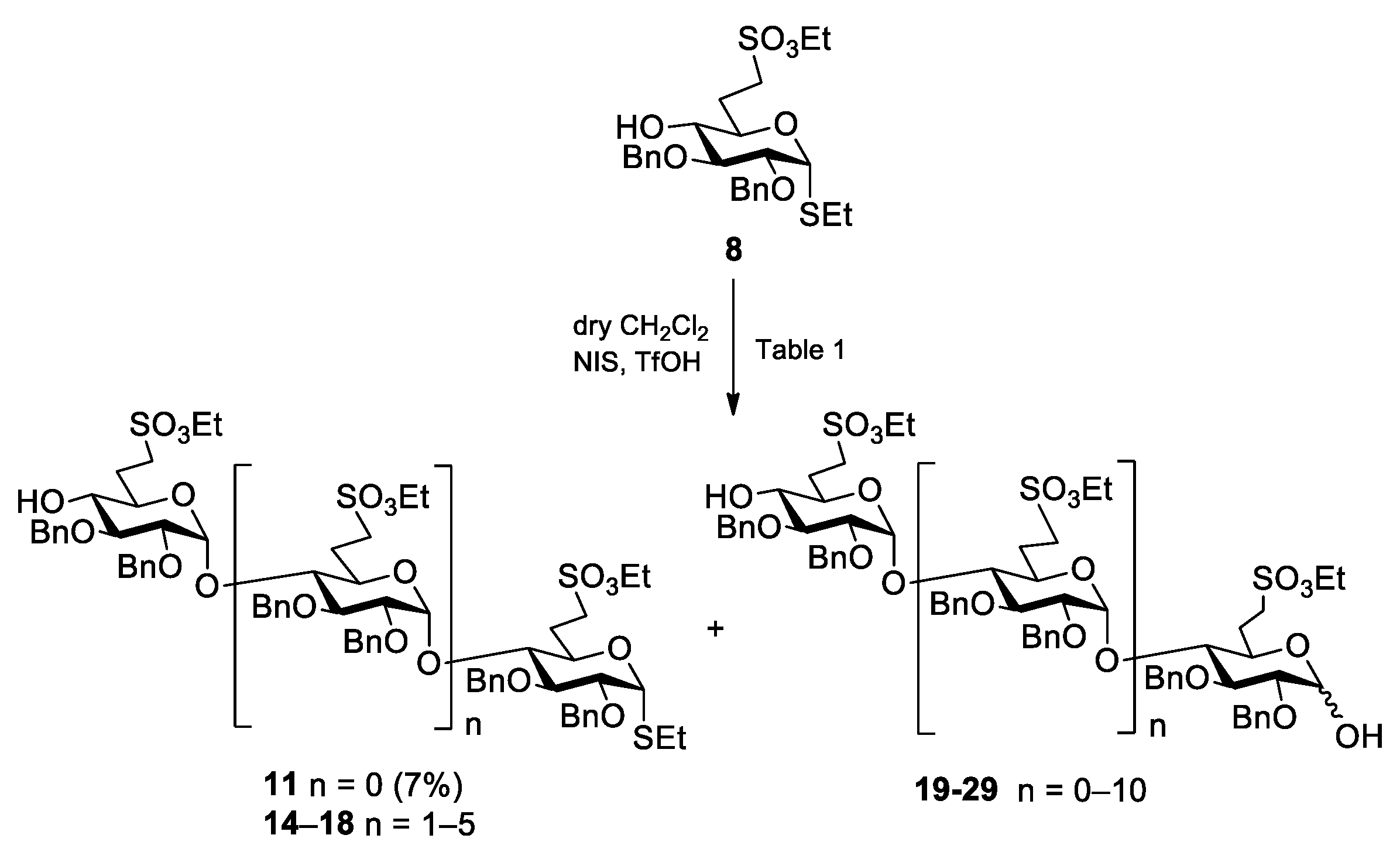
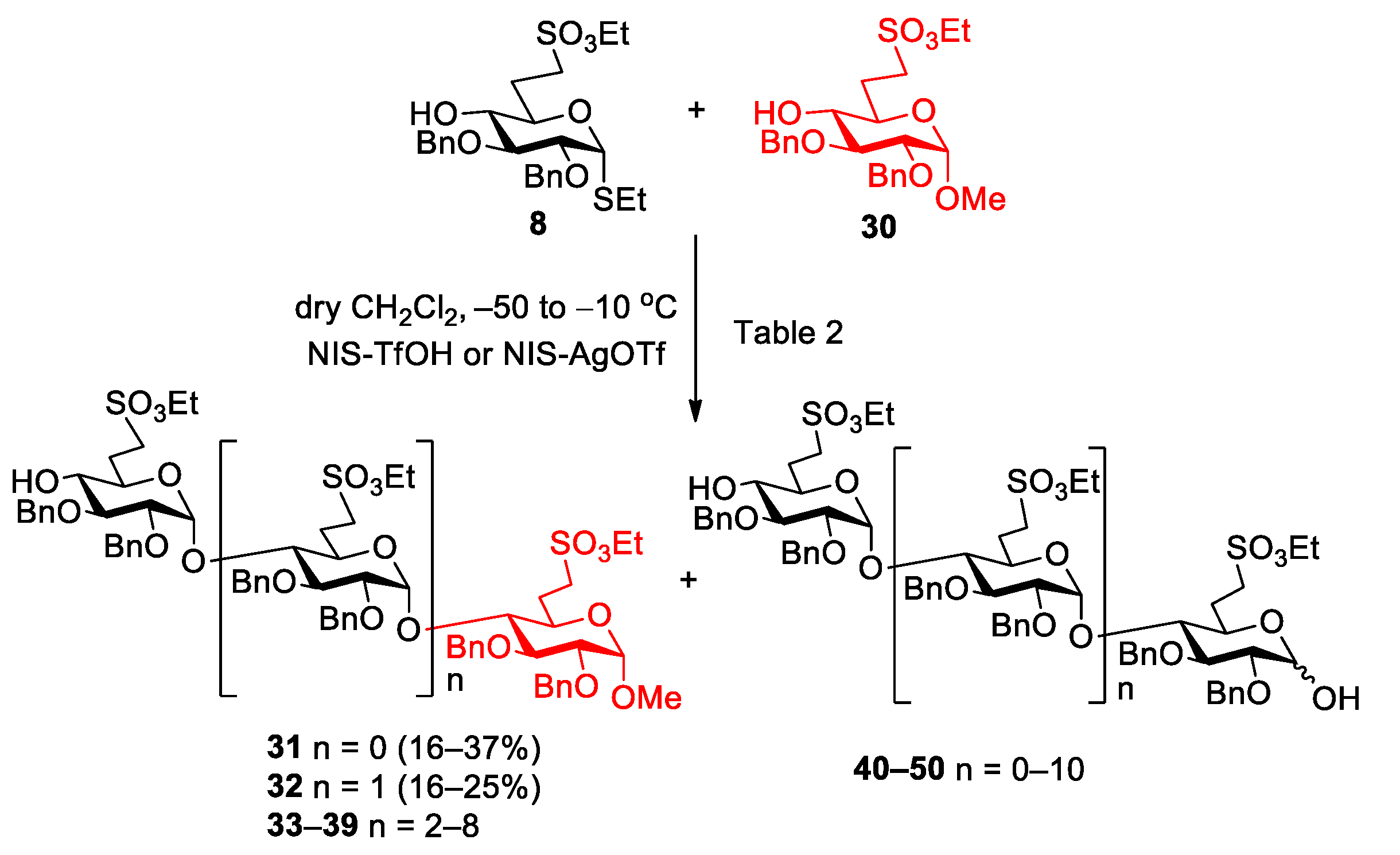
5.2.3. Polymerization Reactions of Compound 8 in the Presence of Acceptor 30 (Table 2, Reactions 1–5)
Methyl [2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (31) and methyl [2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (32).
Reaction 1. To a solution of compound
8 (255 mg, 0.500 mmol) in dry CH
2Cl
2 (18 mL), 4 Å molecular sieves (1.0 g) was added. After 30 min, the stirred mixture was cooled to −50 °C under argon and a mixture of NIS (135 mg, 0.600 mmol, 1.2 equiv.) and TfOH (6 µL, 0.072 mmol, 0.12 equiv.) dissolved in THF (250 µL) was added and the reaction mixture was allowed to warm up to −10 °C. After 2 h, compound
30 (144 mg, 0.300 mmol, 0.6 equiv.) in dry CH
2Cl
2 (2.0 mL) was added and the reaction mixture was stirred for 1 h, at −10 °C. After this, the reaction mixture was diluted with CH
2Cl
2 (100 mL), washed successively with 10% aq. solution of Na
2S
2O
3 (2 × 15 mL), saturated aqueous solution of NaHCO
3 (2 × 15 mL) and H
2O (2 × 15 mL), dried over MgSO
4 and concentrated. The crude product was purified by column chromatography (7:3
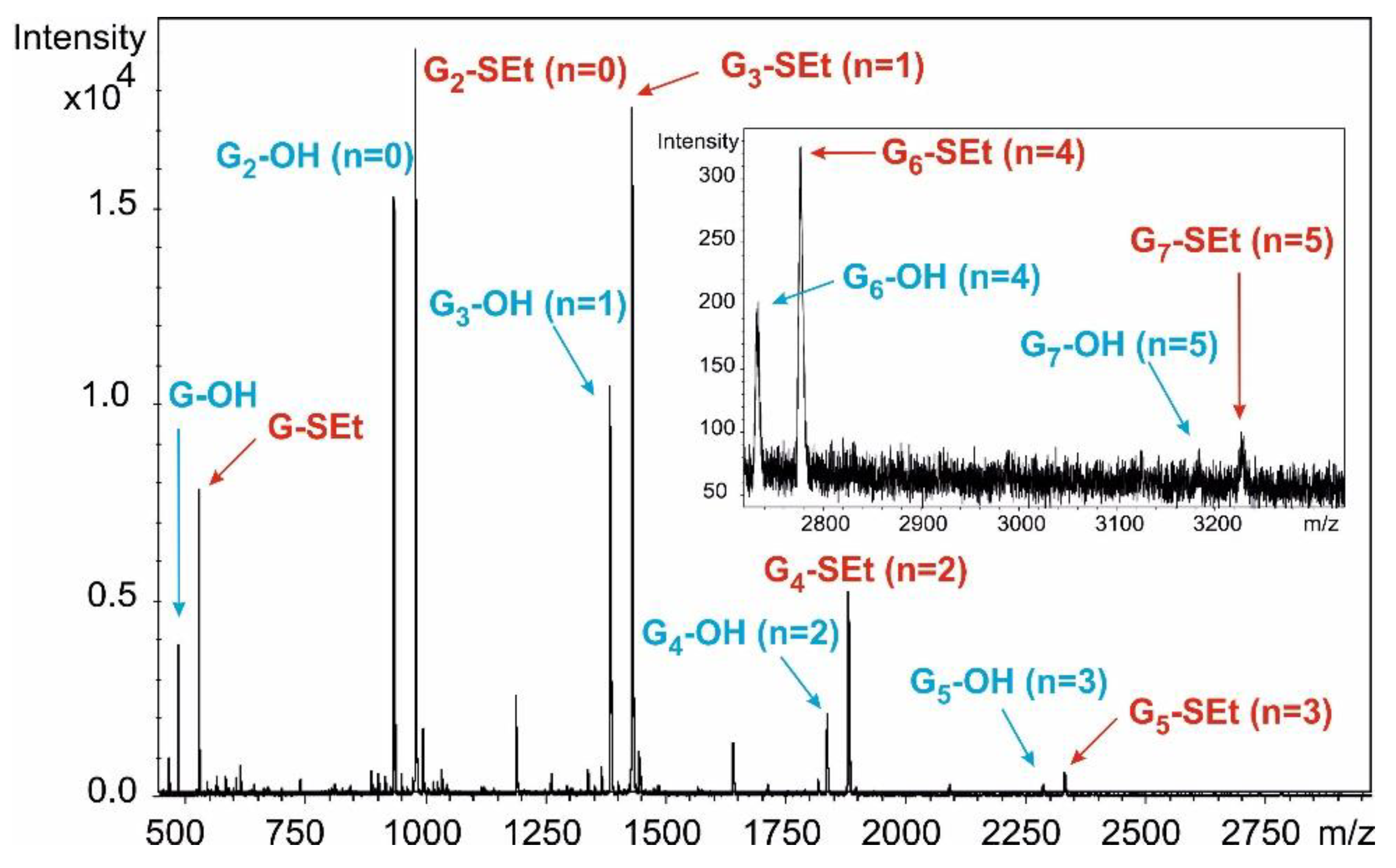
n-hexane/acetone) to give a mixture of hemiacetals as a colourless syrup. (MALDI-TOF MS spectra of the mixture:
Figure S9).
Reaction 2. To a solution of compound
8 (306 mg, 0.600 mmol) and compound
30 (240 mg, 0.500 mmol, 0.8 equiv.) in dry CH
2Cl
2 (18 mL), 4 Å molecular sieves (1.0 g) was added. After 30 min, the stirred mixture was cooled to −50 °C under argon and a mixture of NIS (162 mg, 0.720 mmol, 1.2 equiv.) and TfOH (8.0 µL, 0.086 mmol, 0.12 equiv.) dissolved in THF (300 µL) was added and the reaction mixture was allowed to warm up to −10 °C. After 2.5 h, compound
8 (240 mg, 0.500 mmol, 0.8 equiv.) was added in dry CH
2Cl
2 (1.0 mL). Then, NIS (162 mg, 0.720 mmol, 1.2 equiv.) and TfOH (8.0 µL, 0.086 mmol, 0.12 equiv.) dissolved in THF (300 µL) was added and the reaction mixture was allowed to warm up to 10 °C in 5 h. The reaction mixture was diluted with CH
2Cl
2 (125 mL), washed successively with 10% aq. solution of Na
2S
2O
3 (2 × 20 mL), saturated aqueous solution of NaHCO
3 (2 × 20 mL) and H
2O (2 × 20 mL), dried and concentrated. The crude product was purified by column chromatography (7:3
n-hexane/acetone) to give
31 (171 mg, 37%) as a colourless syrup and
32 (73 mg, 16%) as a colourless syrup. Higher oligomers were detected by MALDI-TOF MS (
Figure S10).
Reaction 3. To a solution of compound
8 (177 mg, 0.346 mmol) and compound
30 (112 mg, 0.231 mmol, 0.6 equiv.) in dry CH
2Cl
2 (9 mL), 4 Å molecular sieves (1.0 g) was added. After 30 min, the stirred mixture was cooled to −50 °C under argon and a mixture of NIS (95 mg, 0.420 mmol, 1.2 equiv.) in dry THF (100 µL) and AgOTf (22 µL, 0.084 mmol, 0.24 equiv.) dissolved in toluene (100 µL) was added and the reaction mixture was allowed to warm up to −10 °C. After 7.5 h, compound
8 (177 mg, 0.346 mmol, 1.0 equiv.) was added in dry CH
2Cl
2 (1.0 mL). Then, NIS (95 mg, 0.420 mmol, 1.2 equiv.) in dry THF (100 µL) and AgOTf (22 µL, 0.084 mmol, 0.24 equiv.) dissolved in toluene (100 µL) was added and the reaction mixture was allowed to warm up to 10 °C in 24 h. The reaction mixture was diluted with CH
2Cl
2 (125 mL), washed successively with 10% aq. solution of Na
2S
2O
3 (2 × 20 mL), saturated aqueous solution of NaHCO
3 (2 × 20 mL) and H
2O (2 × 20 mL), dried over MgSO
4 and concentrated. The crude product was purified by column chromatography (7:3
n-hexane/acetone) to give
31 (35 mg, 16%) as a colourless syrup and
32 (44 mg, 20%) as a colourless syrup. Higher oligomers were detected by MALDI-TOF MS (
Figure S11).
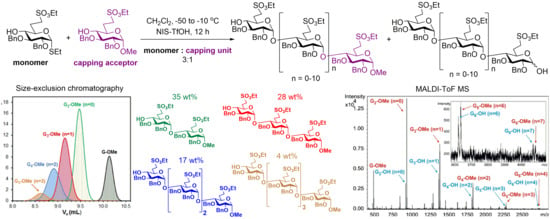
Reaction 4. To a solution of acceptor
30 (240 mg, 0.500 mmol) and donor
8 (765 mg, 1.50 mmol, 3.0 equiv.) in dry CH
2Cl
2 (18 mL), 4 Å molecular sieves (1.0 g) were added. The stirred mixture was cooled to −50 °C under argon. After 30 min at this temperature, a mixture of NIS (405 mg, 1.800 mmol, 1.2 equiv.) and TfOH (19 µL, 0.216 mmol, 0.12 equiv.) dissolved in THF (750 µL) was added and the reaction mixture was allowed to warm up to −10 °C in 12 h. The reaction mixture was diluted with CH
2Cl
2 (200 mL), washed successively with 10% aq. solution of Na
2S
2O
3 (2 × 20 mL), satd. aq. solution of NaHCO
3 (2 × 20 mL) and H
2O (2 × 20 mL), dried over MgSO
4 and concentrated. The crude product was purified by column chromatography (7:3
n-hexane/acetone) to give
31 (153 mg, 33%) as a colourless syrup and
32 (118 mg, 25%) as a colourless syrup. Higher oligomers were detected by MALDI-TOF MS (
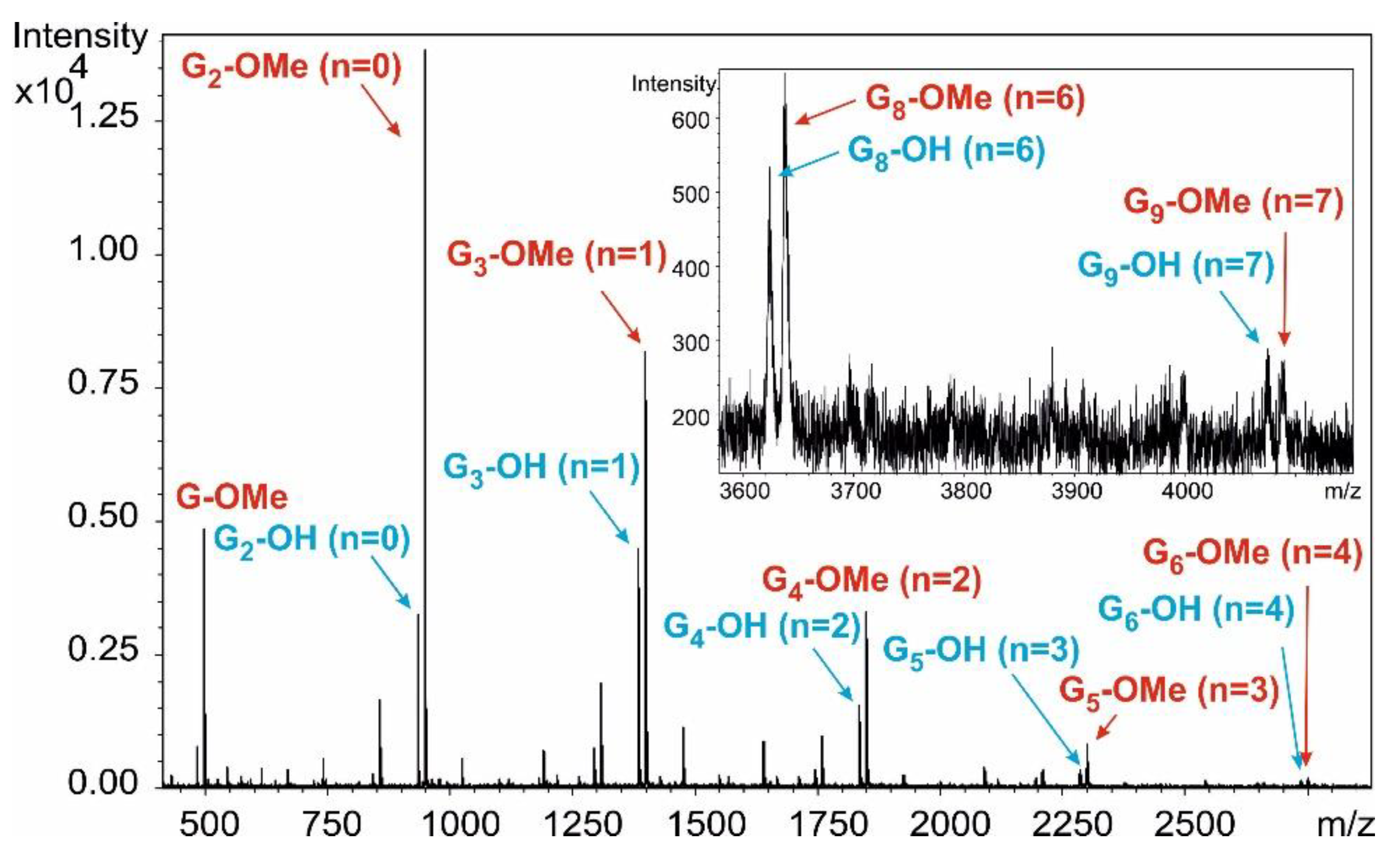
Figure 5).
Reaction 5. To a solution of compound
8 (100 mg, 0.196 mmol, 10 equiv.) and compound
30 (9 mg, 0.019 mmol) in dry CH
2Cl
2 (2.0 mL), 4 Å molecular sieves (200 mg) was added. After 30 min, the stirred mixture was cooled to −50 °C under argon and a mixture of NIS (66 mg, 0.294 mmol, 1.5 equiv.) and TfOH (7.0 µL, 0.088 mmol, 0.3 equiv.) dissolved in THF (100 µL) was added and the reaction mixture was allowed to warm up to 10 °C. After 48 h, the reaction mixture was diluted with CH
2Cl
2 (50 mL), washed successively with 10% aq. solution of Na
2S
2O
3 (2 × 10 mL), saturated aqueous solution of NaHCO
3 (2 × 10 mL) and H
2O (2 × 10 mL), dried and concentrated. Higher oligomers were detected by MALDI-TOF MS (
Figures S12 and S13).
Data of 31: Rf = 0.33 (n-hexane/acetone 7:3); [α]D24 = +48.5 (c = 0.13 in CHCl3); 1H NMR (360 MHz, CDCl3): δ = 7.34–7.15 (m, 20H, Ar-H), 5.66 (d, J1′,2′ = 3.7 Hz, 1H, H-1′), 5.03–4.46 (m, 9H, 4 × PhCH2, H-1), 4.29–4.23 (m, 4H, 2 × SO3CH2CH3), 4.04 (t, J = 9.0 Hz, 1H), 3.81–3.66 (m, 3H) 3.56–3.37 (m, 4H), 3.34 (s, 3H, C-1-OCH3), 3.32–3.06 (m, 4H, H-7a,b, H-7‘a,b), 2.64 (s, 1H, H-4′-OH), 2.46–1.87 (m, 4H, H-6a,b, H-6′a,b), 1.38–1.33 (m, 6H, 2 × SO3CH2CH3) ppm; 13C NMR (90 MHz, CDCl3): δ = 138.8, 138.4, 137.7 (4C, Cq Ar), 128.6–126.5 (20C, Ar), 97.7 (C-1′), 96.8 (C-1), 81.3, 81.0, 80.3, 79.1, 76.7, 73.6, 70.2, 67.5 (8C, skeleton carbons), 75.4, 74.2, 73.4 (4C, 4 × PhCH2), 66.7, 66.4 (2C, 2 × SO3CH2CH3), 55.5 (1C, C-1-OCH3), 46.9, 46.3 (2C, C-7, C-7′), 27.2, 26.5 (2C, C-6, C-6′), 15.2 (2C, 2 × SO3CH2CH3) ppm; MALDI-TOF (positive ion): m/z calcd for C47H60NaO15S2 [M + Na]+: 951.33; found: 951.35.
Data of 32: Rf = 0.24 (n-hexane/acetone 7:3); [α]D24 = +51.8 (c = 0.11 in CHCl3); 1H NMR (360 MHz, CDCl3): δ = 7.35–7.08 (m, 30H, Ar-H), 5.56 (d, J1′,2′ = 3.4 Hz, 1H, H-1′), 5.34 (d, J1″,2″ = 3.4 Hz, 1H, H-1″), 5.01–4.39 (m, 13H, 6 × PhCH2, H-1), 4.30–4.19 (m, 6H, 3 × SO3CH2CH3), 4.09–3.44 (m, 12H), 3.36 (s, 3H, C-1-OCH3), 3.34–3.04 (m, 6H, H-7a,b, H-7‘a,b, H-7″a,b), 2.81–2.73 (m, 1H, H-4″-OH), 2.52–1.86 (m, 6H, H-6a,b, H-6′a,b, H-6″a,b), 1.39–1.25 (m, 9H, 3 × SO3CH2CH3) ppm; 13C NMR (90 MHz, CDCl3): δ = 138. 9, 138.7, 138.6, 137.9, 137.7, 137.5 (6C, Cq Ar), 128.7–126.6 (30C, Ar), 98.0, 97.8 (2C, C-1′, C1″), 96.6 (C-1), 81.3, 80.8, 80.4, 80.3, 79.3, 79.2, 79.0, 74.0, 70.6, 69.4, 67.8 (12C, skeleton carbons), 75.4, 75.3, 74.4, 73.5, 73.4 (6C, 6 × PhCH2), 67.0, 66.8, 66.6 (3C, 3 × SO3CH2CH3), 55.6 (1C, C-1-OCH3), 47.0, 46.6, 46.3 (3C, C-7, C-7′, C-7″), 27.5, 27.2, 26.7 (3C, C-6, C-6′, C-6″), 15.3, 15.2 (3C, 3 × SO3CH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C70H88O22S3 + 2Na]2+: 711.2357; found: 711.2357.
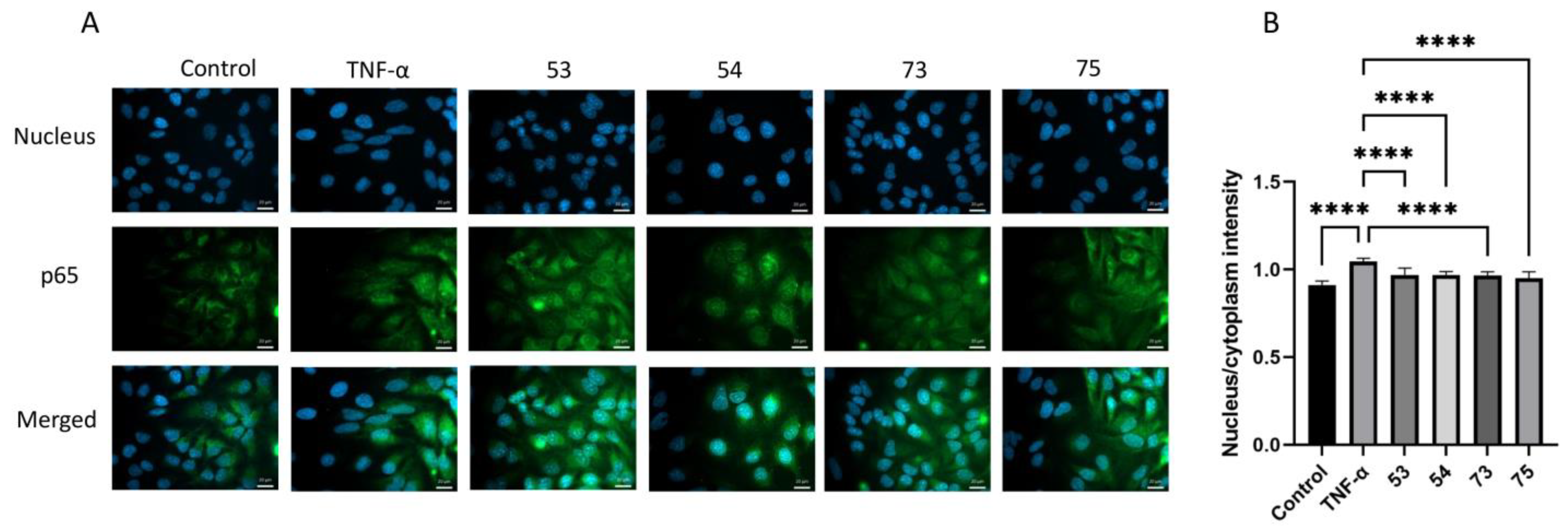
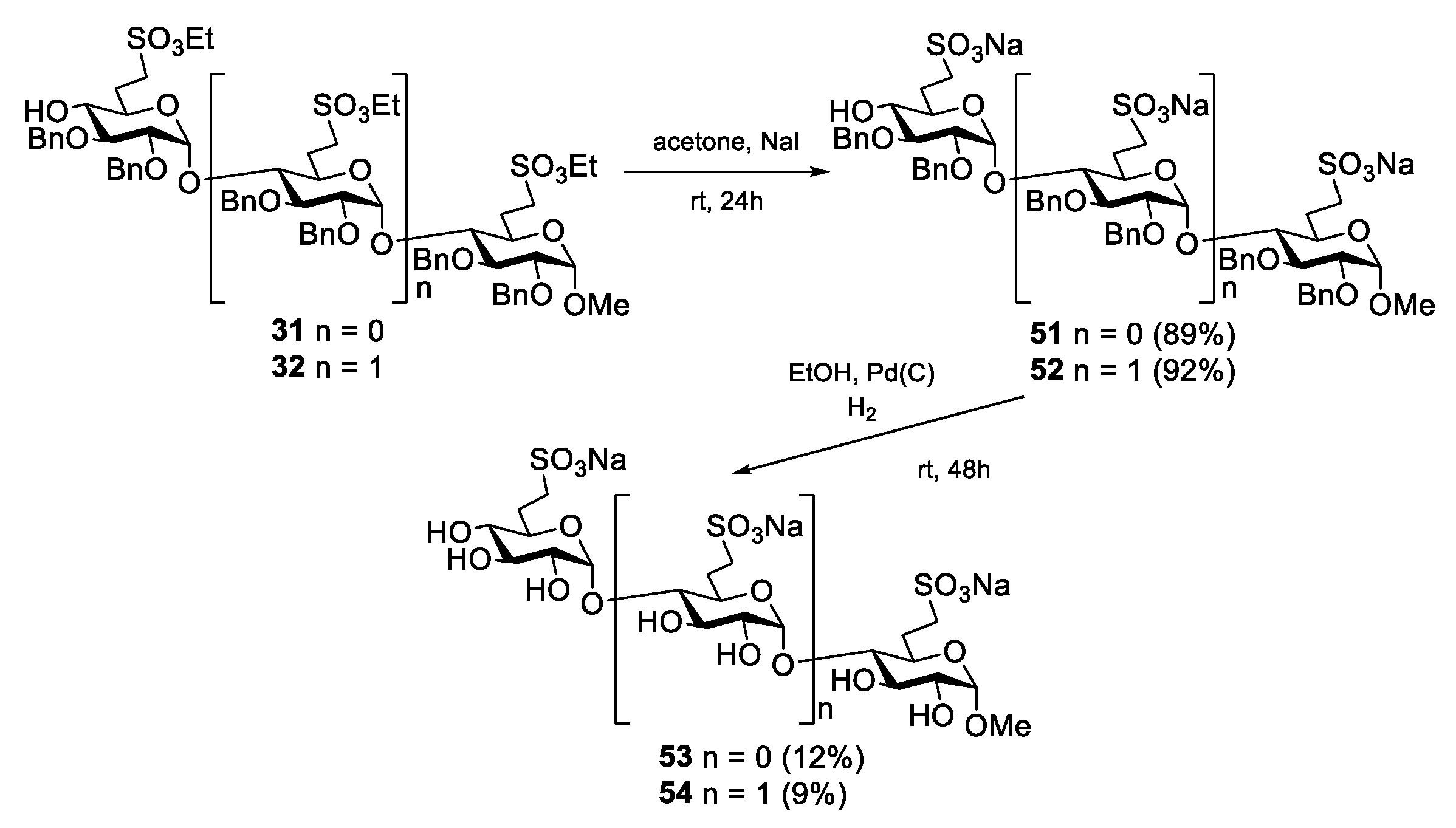
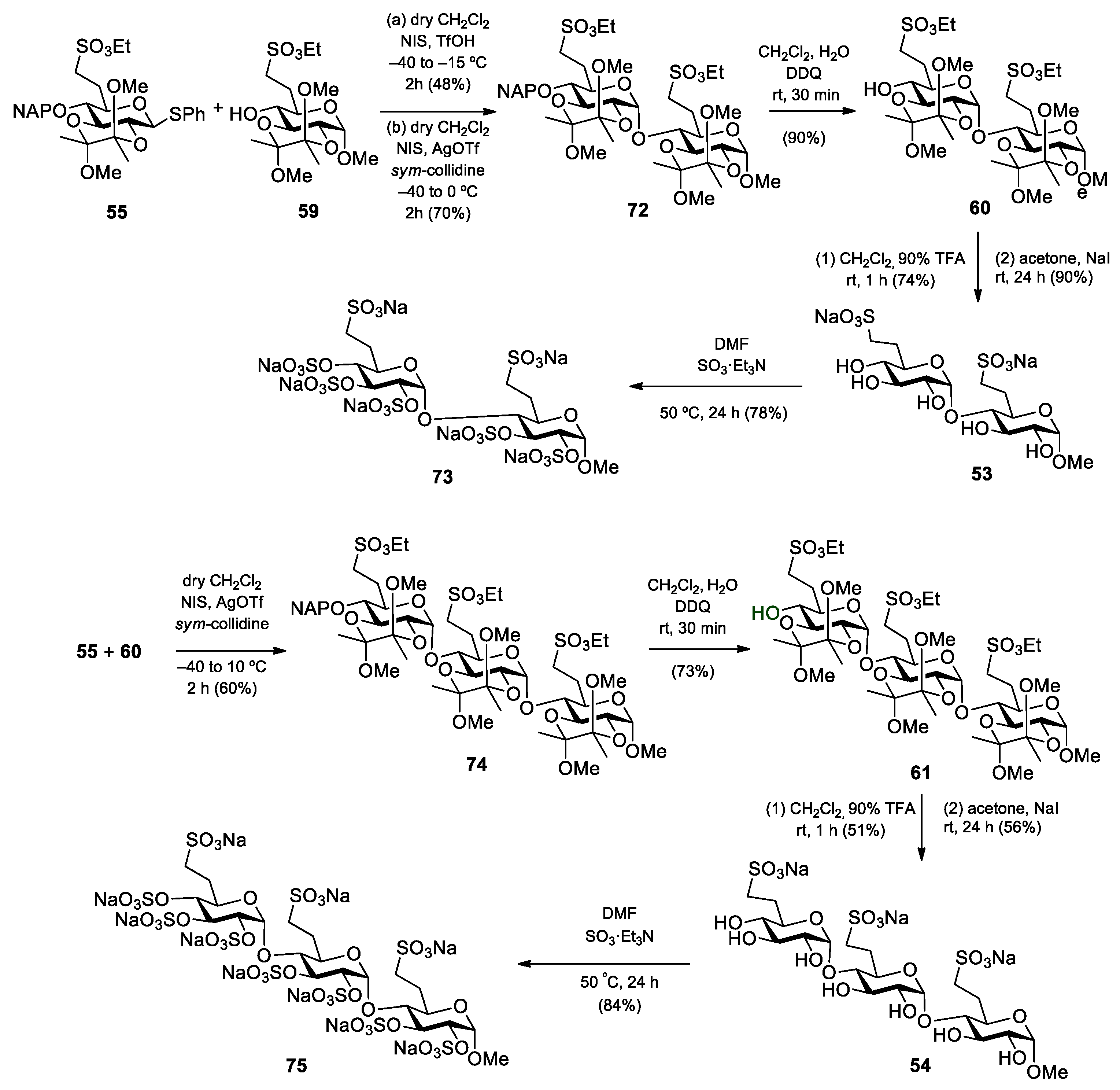
Methyl (6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl)-(1→4)-(6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranoside disodium salt (53).
Method I: Compound 31 (396 mg, 0.158 mmol) was converted to disodium salt according to general method C. The residue was purified by column chromatography on Sephadex LH-20 (MeOH) to give 51 (347 mg, 89%) as a white solid. Rf = 0.43 (CH2Cl2/MeOH 8:2); [α]D24 = +56.2 (c = 0.13 in CHCl3); 1H NMR (360 MHz, CDCl3 + CD3OD): δ = 7.34–7.10 (m, 20H, Ar-H), 5.66 (s, 1H, H-1′), 4.93–4.41 (m, 9H, 4 × PhCH2, H-1), 4.01–3.55 (m, 8H), 3.36 (s, 3H, C-1-O-CH3), 3.33–3.15 (m, 4H, H-7a,b, H-7‘a,b), 2.95 (s, 1H, H-4′-OH), 2.49–1.95 (m, 4H, H-6a,b, H-6′a,b) ppm; 13C NMR (90 MHz, CDCl3 + CD3OD): δ = 138.5, 138.2, 137.6, 137.3 (4C, Cq Ar), 128.4–125.5 (20C, Ar), 97.0 (C-1′), 96.7 (C-1), 81.2, 80.9, 79.9, 78.7, 74.1, 71.1, 67.8 (8C, skeleton carbons), 74.9, 73.4, 72.8, 72.4 (4C, 4 × PhCH2), 54.3 (1C, C-1-OCH3), 46.7, 46.1 (2C, C-7, C-7′), 27.4, 26.7 (2C, C-6, C-6′) ppm; MALDI-TOF (positive ion): m/z calcd for C43H50Na3O15S2 [M + Na]+: 939.23; found: 939.31. The disodium salt (51) (300 mg, 0.327 mmol) was dissolved in EtOH/AcOH (96%, 29:1, 15 mL), Pd/C (10%, 235 mg) was added and the mixture was stirred in an autoclave under a H2 atmosphere (10 bar) for 48 h. The catalyst was filtered through a pad of Celite and the filtrate was concentrated. The crude product was treated with Dowex ion-exchange resin (Na+ form) and then purified by column chromatography on Sephadex LH-20 (MeOH) to give compound 53 (21 mg, 12%) as a white solid.
Method II: To a solution of compound 60 (55 mg, 0.069 mmol) in CH2Cl2 (1.0 mL), 90% F3CCOOH (500 µL) was added. After 1 h, the mixture was diluted with toluene and concentrated. The residue was purified by column chromatography on Sephadex LH-20 (MeOH) to give the pentaol (25 mg, 74%) as a white solid. Rf = 0.53 (CH2Cl2/MeOH 8:2); [α]D24 = +30.0 (c = 0.11 in MeOH); MALDI-TOF (positive ion): m/z calcd for C19H36NaO15S2 [M + Na]+: 591.19; found: 591.30. The pentaol (20 mg, 0.035 mmol) was converted to 53 according to general method C in acetone (1.0 mL) and H2O (100 µL). The residue was purified by column chromatography on Sephadex LH-20 (MeOH) to give the disodium salt 53 (17 mg, 90%) as a white solid. Rf = 0.41 (CH2Cl2/MeOH/H2O 7:6:1); [α]D24 = +60.4 (c = 0.24 in MeOH); 1H NMR (400 MHz, D2O): δ = 5.58 (s, 1H, H-1′), 4.67 (d, 1H, H-1), 4.35–4.24 (m, 5H, 2 × SO3CH2CH3, H-3), 3.86–3.80 (m, 2H, H-3′, H-5), 3.74–3.56 (m, 4H, H-2, H-2′, H-4, H-5′), 3.43–3.40 (m, 1H, H-4′), 3.38, 3.36, 3.29, 3.26, 3.21 (5 × s, 15H, C-1-OCH3, 4 × BDA-OCH3), 3.23–3.19 (m, 3H, 2 × H-7a, H-7b), 3.12–3.07 (m, 1H, H-7b), 2.66–2.61 (m, 1H, H-4′-OH), 2.42–2.36 (m, 2H, 2 × H-6a), 2.05–1.87 (m, 2H, 2 × H-6b), 1.44–1.19 (m, 18H, 2 × SCH2CH3, 4 × BDA-CH3) ppm; 13C NMR (100 MHz, D2O): δ = 100.0 (C-1′), 99.9 (C-1), 80.8 (C-4), 74.7 (C-3), 73.9 (C-4′), 73.4 (C-3′), 72.7 (C-2′), 72.1 (C-2), 71.3 (C-5′), 68.9 (C-5), 56.1 (1C, C-1-OCH3), 47.9 (2C, C-7, C-7′), 27.8, 27.3 (2C, C-6, C-6′) ppm; UHR ESI-QTOF (positive ion): m/z calcd for C15H26Na3O15S2 [M + Na]+: 579.0401; found: 579.0402.
Methyl (6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl)-(1→4)-(6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl)]-(1→4)-(6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranoside) trisodium salt (54).
Method I: Compound 32 (220 mg, 0.159 mmol) was converted to trisodium salt according to general method C. The residue was purified by column chromatography on Sephadex LH-20 (MeOH) to give the trisodium salt 52 (201 mg, 92%) as a white solid. Rf = 0.51 (CH2Cl2/MeOH 8:2); MALDI-TOF (positive ion): m/z calcd for C64H73Na4O22S3 [M + Na]+: 1381.33; found: 1381.22. The trisodium salt 52 (201 mg, 0.148 mmol) was dissolved in EtOH/AcOH (96%, 29:1, 5 mL), Pd/C (10%, 50 mg) was added and the mixture was stirred in an autoclave under a H2 atmosphere (10 bar) for 48 h. The catalyst was filtered through a pad of Celite and the filtrate was concentrated. The crude product was treated with Dowex ion-exchange resin (Na+ form) and then purified by column chromatography on Sephadex LH-20 (MeOH) to give compound 54 (11 mg, 9%) as a white solid.
Method II: To a solution of compound 61 (120 mg, 0.101 mmol) in CH2Cl2 (1.5 mL), 90% F3CCOOH (731 µL) was added. After 1 h, the mixture was diluted with toluene and concentrated. The residue was purified by column chromatography (85:15 CH2Cl2/MeOH) to give the heptaol (46 mg, 51%) as a white solid. Rf = 0.13 (CH2Cl2/MeOH 9:1); [α]D24 = +96.4 (c = 0.11 in MeOH); 1H NMR (400 MHz, CD3OD): δ = 5.16 (d, J1′,2′ =3.6 Hz, 1H, H-1′), 5.13 (d, J1″,2″ = 3.6 Hz, 1H, H-1″), 4.65 (d, J1,2 = 3.7 Hz, 1H, H-1), 4.36–4.27 (m, 6H, 3 × SO3CH2CH3), 3.87–3.44 (m, 12H, H-2, H-2′, H-2″, H-3, H-3′, H-3″, H-4, H-4′, H-4″, H-5, H-5′, H-5″), 3.40 (s, 3H, C-1-OCH3), 3.38–3.24 (m, 6H, 3 × H-7a,b), 2.43–2.36 (m, 3H, 3 × H-6a), 1.99–1.89 (m, 3H, 3 × H-6b), 1.42–1.36 (m, 9H, 3 × SCH2CH3) ppm; 13C NMR (100 MHz, CD3OD): δ = 103.3 (C-1), 101.1 (C-1′), 101.0 (C-1″), 85.9, 75.1, 74.7, 74.5, 74.4, 74.2, 73.6, 73.0, 72.2, 70.4, 68.9 (12C, skeleton carbons), 68.5, 68.4, 68.2 (3C, 3 × SO3CH2CH3), 56.0 (1C, C-1-OCH3), 47.2, 47.0 (3C, 3 × C-7), 27.6, 27.4, 27.3 (3C, 3 × C-6), 15.6, 15.5, 15.4 (3C, 3 × SO3CH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for C28H52NaO22S3 [M + Na]+: 859.2005; found: 859.2002. The heptaol (40 mg, 0.048 mmol) was converted to 54 according to general method C in acetone (1.5 mL) and H2O (250 µL). The residue was purified by column chromatography on Sephadex LH-20 (MeOH) to give the trisodium salt 54 (22 mg, 56%) as a white solid. Rf = 0.45 (CH2Cl2/MeOH/H2O 7:6:1); [α]D24 = +123.0 (c = 0.10 in H2O); 1H NMR (400 MHz, D2O): δ = 5.53 (d, J1″,2″ = 3.7 Hz, 1H, H-1″), 5.48 (d, J1′,2′ = 3.7 Hz, 1H, H-1′), 4.80 (d, 1H, H-1), 3.98–3.87 (m, 4H, H-3, H-3′, H-5, H-5′), 3.80 (t, J = 7.5 Hz, 1H, H-5″), 3.71 (t, J = 9.5 Hz, 1H, H-3″), 3.63–3.60 (m, 2H, H-2, H-2′), 3.56 (dd, J = 10.0 Hz, J = 3.8 Hz, 1H, H-2″), 3.50–3.45 (m, 2H, H-4, H-4′), 3.43 (s, 3H, C-1-O-CH3), 3.24 (t, J = 9.4 Hz, 1H, H-4″), 3.16–3.01 (m, 6H, H-7a,b, H-7‘a,b, H-7″a,b), 2.39–2.28 (m, 3H, H-6a, H-6′a, H-6″a), 1.97–1.90 (m, 3H, H-6b, H-6′b, H-6″b) ppm; 13C NMR (100 MHz, D2O): δ = 99.9 (C-1), 99.6 (C-1″), 99.5 (C-1′), 80.6 (C-4), 79.9 (C-4′), 74.6 (C-3), 74.2 (C-3′), 73.9 (C-4″), 73.3 (C-3″), 72.7 (C-2″), 72.6, 72.2 (2C, C-2, C-2′), 71.7 (C-5″), 70.2 (C-5′), 68.9 (C-5), 56.1 (1C, C-1-OCH3), 47.9, 47.8 (3C, C-7, C-7′, C-7″), 28.1, 27.8 (2C, C-6, C-6′), 27.3 (1C, C-6″) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C22H37Na3O22S3 + Na]+: 841.0524; found: 841.0526.
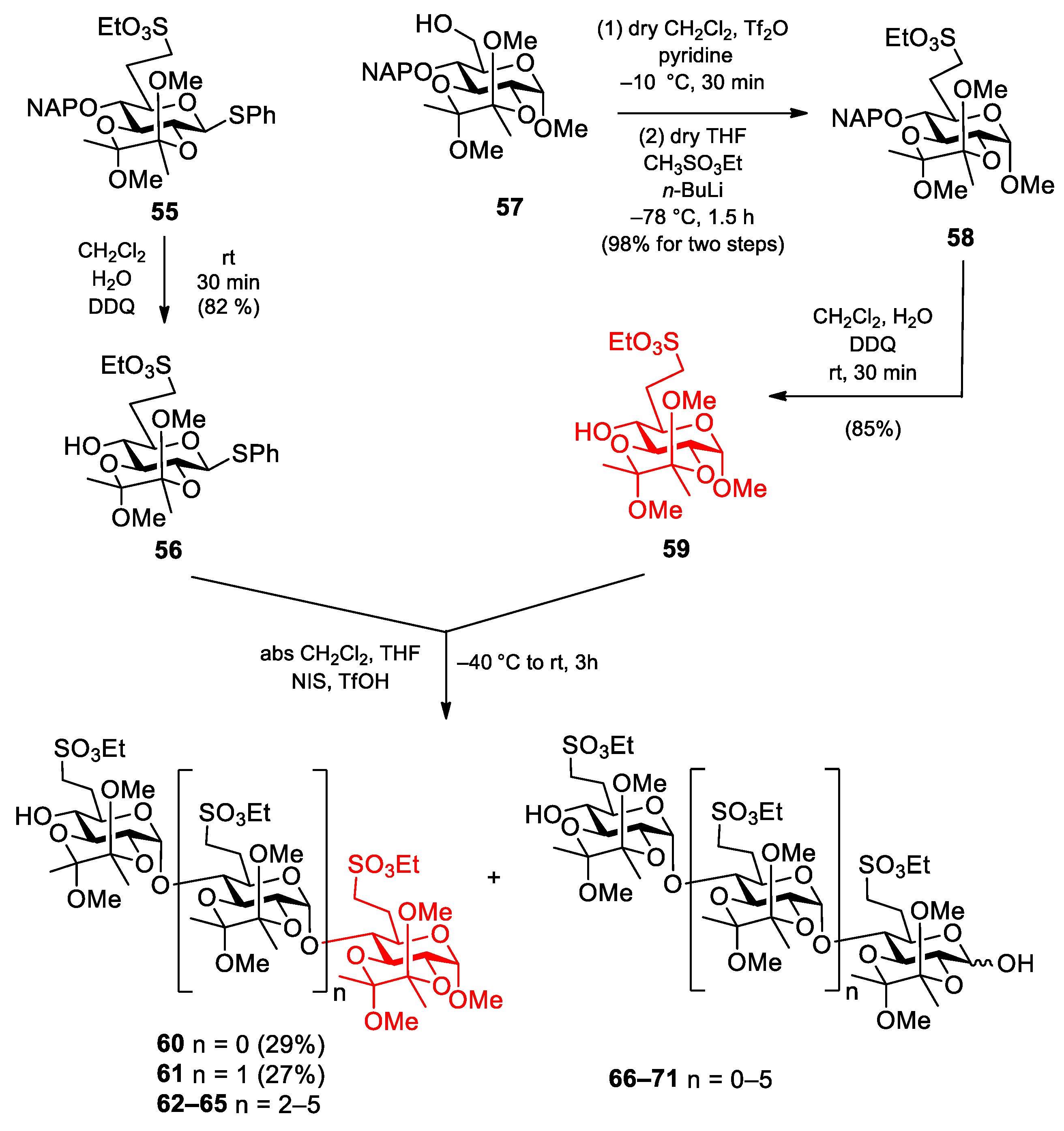
Phenyl 2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-1-thio-β-d-glucopyranoside (56). Compound 55 (900 mg, 1.400 mmol) was converted to 56 according to general method B. The crude product was purified by column chromatography (1:1 n-hexane/EtOAc) to give 56 (574 mg, 82%) as a colourless syrup. Rf = 0.57 (n-hexane/EtOAc 1:1); [α]D24 = −122.1 (c = 0.05 in CHCl3); 1H NMR (500 MHz, CDCl3): δ = 7.50–7.26 (m, 5H, Ar-H), 4.76 (d, J1,2 = 9.7 Hz, 1H, H-1), 4.21 (p, J = 7.2 Hz, 2H, SO3CH2CH3), 3.69 (t, J = 9.2 Hz, 1H, H-3), 3.60 (t, J = 9.6 Hz, 1H, H-2), 3.47 (t, J = 9.0 Hz, 1H, H-4), 3.40 (t, J = 9.0 Hz, 1H, H-5), 3.28, 3.23 (2 × s, 6H, 2 × BDA-OCH3), 3.22–3.18 (m, 1H, H-7a), 3.14–3.08 (m, 1H, H-7b), 2.64 (s, 1H, H-4-OH), 2.49–2.42 (m, 1H, H-6a), 2.02–1.96 (m, 1H, H-6b), 1.38–1.33 (m, 9H, SCH2CH3, 2 × BDA-CH3) ppm; 13C NMR (125 MHz, CDCl3): δ = 132.7 (1C, Cq Ar), 132.3, 129.0, 127.8 (5C, Ar), 100.2, 99.8 (2C, 2 × Cq BDA), 84.9 (1C, C-1), 77.6 (1C, C-5), 74.3 (1C, C-3), 71.0 (1C, C-4), 68.3 (1C, C-2), 66.3 (1C, SO3CH2CH3), 48.3, 48.1 (2C, 2 × BDA-OCH3), 46.4 (1C, C-7), 26.3 (1C, C-6), 17.7 (2C, 2 × BDA-CH3), 15.2 (1C, SO3CH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C21H32O9S2 + Na]+: 515.1380; found: 515.1379.
Methyl 2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-4-O-(2-naphthyl)methyl-α-
d-glucopyranoside (
58). To a solution of
57 [
45] (2.59 g, 5.780 mmol) in dry CH
2Cl
2 (16 mL), dry pyridine (1.6 mL) was added. The stirred mixture was cooled to −10 °C under argon and trifluoromethanesulfonic anhydride (1.36 mL, 8.09 mmol, 1.4 equiv.) dissolved in CH
2Cl
2 (3.5 mL) was added. After the complete disappearance of the starting material (30 min), the reaction mixture was diluted with CH
2Cl
2 (150 mL) and washed successively with H
2O (2 × 25 mL), 1 M HCl (2 × 15 mL), H
2O (2 × 25 mL), satd. aq. solution of NaHCO
3 (2 × 15 mL) and H
2O (15 mL). The organic phase was dried over MgSO
4 and concentrated at 30 °C. The crude product was used for further reaction without purification. A solution of methanesulfonic acid ethyl ester (1.19 mL, 11.56 mmol, 2.0 equiv.) in dry THF (36 mL) was cooled to −80 °C under argon and 2.5 M
n-BuLi (4.63 mL, 11.56 mmol, 2.0 equiv.) in
n-hexane was added. After stirring at −78 °C for 30 min, a solution of the triflate (3.35 g, 5.78 mmol) in dry THF (18 mL) was added dropwise. The reaction mixture was stirred for 1.5 h while its temperature was raised to −20 °C. The stirred mixture was quenched by the addition of satd. aq. solution of NH
4Cl (40 mL) and diluted with EtOAc (200 mL). The organic phase was washed successively with satd. aq. solution of NH
4Cl (2 × 15 mL), H
2O (2 × 20 mL), satd. aq. solution of NaCl (2 × 15 mL), dried over MgSO
4 and concentrated. The crude product was purified by column chromatography (1:1
n-hexane/EtOAc) to give
58 (3.15 g, 98% for two steps) as a colourless syrup.
Rf = 0.54 (
n-hexane/EtOAc 1:1); [α]
D24 = +32.8 (
c = 0.25 in CHCl
3);
1H NMR (400 MHz, CDCl
3):
δ = 7.84–7.46 (m, 7H, Ar-H), 5.15 (d,
Jgem = 11.3 Hz, 1H, NpC
H2a), 4.84 (d,
Jgem = 11.3 Hz, 1H, NpC
H2b), 4.68 (d,
J1,2 = 3.5 Hz, 1H, H-1), 4.31–4.28 (m, 3H, SO
3C
H2CH
3, H-3), 3.77 (dd,
J = 10.3 Hz,
J = 3.6 Hz, 1H, H-2), 3.71 (dt,
J = 9.1 Hz,
J = 2.9 Hz, 1H, H-5), 3.38 (t,
J = 9.2 Hz, 1H, H-4), 3.37, 3.32, 3.28 (3 × s, 9H, C-1-OC
H3, 2 × BDA-OC
H3), 3.25–3.05 (m, 2H, H-7a,b), 2.42–2.40 (m, 1H, H-6a), 2.03–1.89 (m, 1H, H-6b), 1.42–1.28 (m, 9H, SCH
2C
H3, 2 × BDA-C
H3) ppm;
13C NMR (100 MHz, CDCl
3):
δ = 135.5, 133.2, 133.0 (3C, C
q Ar), 128.3–126.0 (7C, Ar), 99.9, 99.4 (2C, 2 × C
q BDA), 97.7 (C-1), 78.4 (1C, C-4), 70.5 (1C, C-3), 68.7 (1C, C-5), 68.3 (1C, C-2), 74.9 (1C, Np
CH
2), 66.2 (1C, SO
3CH
2CH
3), 55.1 (1C, C-1-O
CH
3), 48.0, 47.9 (2C, 2 × BDA-O
CH
3), 46.7 (1C, C-7), 26.0 (1C, C-6), 18.0, 17.7 (2C, 2 × BDA-
CH
3), 15.0 (1C, SO
3CH
2CH
3) ppm; UHR ESI-QTOF (positive ion):
m/
z calcd for [C
27H
38O
10S + Na]
+: 577.2078; found: 577.2079.
Methyl 2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (59). Compound 58 (3.15 g, 5.680 mmol) was converted to 59 according to general method B. The crude product was purified by column chromatography (7:3 n-hexane/acetone) to give 59 (1.99 g, 85%) as a colourless syrup. Rf = 0.31 (n-hexane/acetone 7:3); [α]D24 = +92.4 (c = 0.45 in CHCl3); 1H NMR (400 MHz, CDCl3): δ = 4.70 (d, J1,2 = 3.5 Hz, 1H, H-1), 4.31 (q, J = 7.1 Hz, 2H, SO3CH2CH3), 3.97 (dd, J = 10.0 Hz, J = 9.5 Hz, 1H, H-3), 3.69 (dd, J = 10.4 Hz, J = 3.6 Hz, 1H, H-2), 3.66 (dt, J = 9.2 Hz, J = 3.0 Hz, 1H, H-5), 3.44 (t, J = 9.1 Hz, 1H, H-4), 3.40, 3.29, 3.26 (3 × s, 9H, C-1-OCH3, 2 × BDA-OCH3), 3.37–3.31 (m, 1H, H-7a), 3.21–3.14 (m, 1H, H-7a), 2.88 (d, J = 2.1 Hz, 1H, H-4-OH), 2.47–2.39 (m, 1H, H-6a), 2.05–1.96 (m, 1H, H-6b), 1.42 (t, J = 7.1 Hz, 3H, SCH2CH3), 1.34, 1.31 (2 × s, 6H, 3 × BDA-CH3) ppm; 13C NMR (100 MHz, CDCl3): δ = 99.9, 99.5 (2C, 2 × Cq BDA), 98.0 (1C, C-1), 71.5 (1C, C-4), 69.5 (1C, C-5), 69.2 (1C, C-3), 68.2 (1C, C-2), 66.2 (1C, SO3CH2CH3), 55.2 (1C, C-1-OCH3), 48.0, 47.9 (2C, 2 × BDA-OCH3), 46.7 (1C, C-7), 25.8 (1C, C-6), 17.7, 17.6 (2C, 2 × BDA-CH3), 15.1 (1C, SO3CH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C16H30O10S + Na]+: 437.1452; found: 437.1453.
Methyl [2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (60) and methyl [2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (61).
To a solution of acceptor
59 (140 mg, 0.338 mmol) and donor
56 (500 mg, 1.015 mmol, 3.0 equiv.) in dry CH
2Cl
2 (18 mL), 4 Å molecular sieves (1.0 g) were added. The stirred mixture was cooled to −40 °C under argon. After 30 min at this temperature, a mixture of NIS (320 mg, 1.421 mmol, 1.4 equiv.) and TfOH (31 µL, 0.355 mmol) dissolved in THF (750 µL) was added and the reaction mixture was allowed to warm up to rt in 2.5 h. The reaction mixture was diluted with CH
2Cl
2 (200 mL), washed successively with 10% aq. solution of Na
2S
2O
3 (2 × 25 mL), satd. aq. solution of NaHCO
3 (2 × 25 mL) and H
2O (2 × 25 mL), dried over MgSO
4 and concentrated. The crude product was purified by column chromatography (1:1
n-hexane/EtOAc) to give
60 (78 mg, 29%) as a colourless syrup and
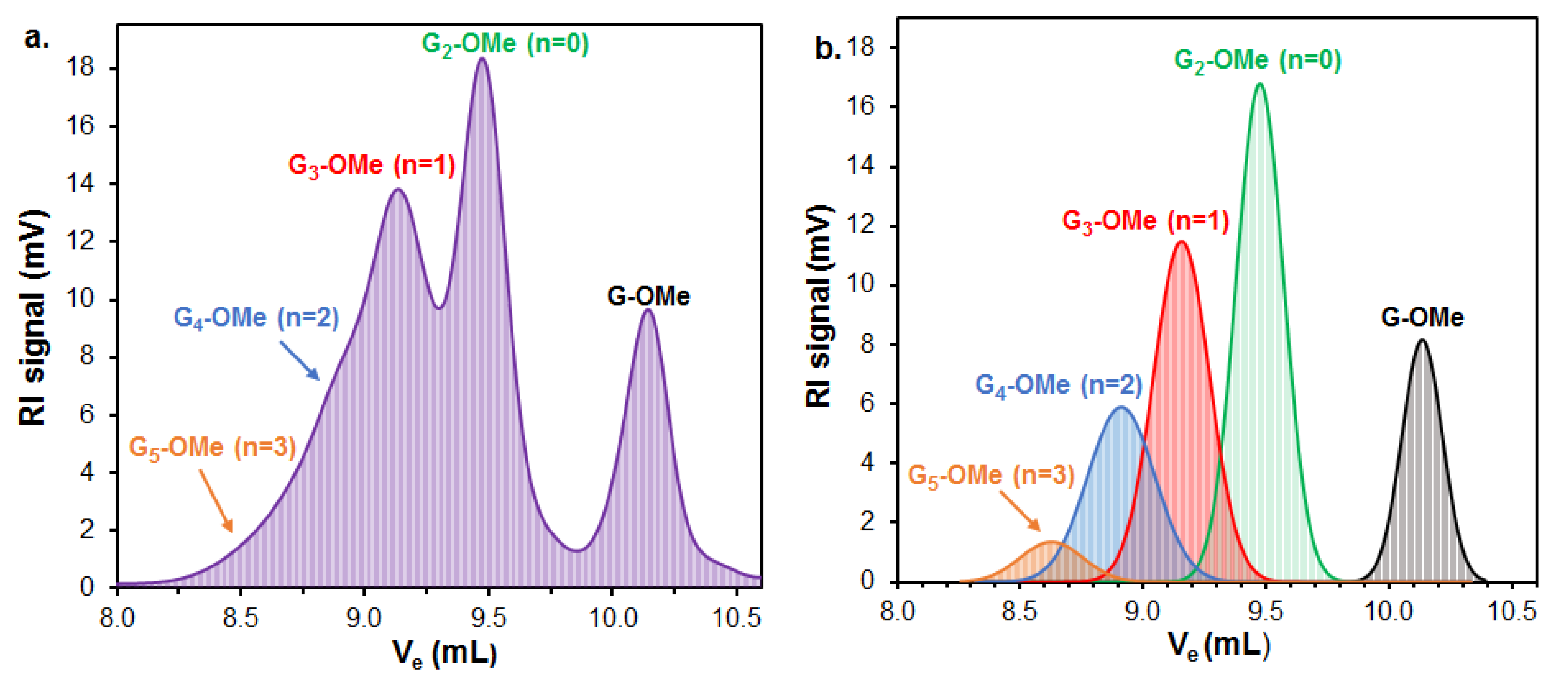
61 (107 mg, 27%) as a colourless syrup. (Higher oligomers were detected by MALDI-TOF MS, see
Figure S14).
Data of disaccharide 60: Rf = 0.19 (n-hexane/EtOAc 1:1); [α]D24 = −14.6 (c = 0.11 in CHCl3); 1H NMR (400 MHz, CDCl3): δ = 5.58 (s, 1H, H-1′), 4.67 (s, 1H, H-1), 4.35–4.24 (m, 5H, 2 × SO3CH2CH3, H-3), 3.86–3.80 (m, 2H, H-3′, H-5), 3.74–3.56 (m, 4H, H-2, H-2′, H-4, H-5′), 3.43–3.40 (m, 1H, H-4′), 3.38, 3.36, 3.29, 3.26, 3.21 (5 × s, 15H, C-1-OCH3, 4 × BDA-OCH3), 3.23–3.19 (m, 3H, 2 × H-7a, H-7b), 3.13–3.06 (m, 1H, H-7b), 2.72–2.67 (m, 1H, H-4′-OH), 2.42–2.36 (m, 2H, 2 × H-6a), 2.05–1.87 (m, 2H, 2 × H-6b), 1.44–1.19 (m, 18H, 2 × SCH2CH3, 4 × BDA-CH3) ppm; 13C NMR (100 MHz, CDCl3): δ = 99.8, 99.7, 99.4, 99.1 (4C, 4 × Cq BDA), 97.8 (1C, C-1), 96.9 (1C, C-1′), 75.3 (1C, C-4), 71.4 (1C, C-4′), 70.5 (1C, C-5′), 69.8 (1C, C-3), 68.9 (1C, C-5), 68.3 (1C, C-2), 67.8 (1C, C-3′), 67.6 (1C, C-2′), 66.7, 66.3 (2C, 2 × SO3CH2CH3), 55.2 (1C, C-1-OCH3), 48.0, 47.9, 47.6 (4C, 4 × BDA-OCH3), 46.7, 46.3 (2C, 2 × C-7), 27.1, 26.2 (2C, 2 × C-6), 17.8, 17.7, 17.3 (4C, 4 × BDA-CH3), 15.1, 15.0 (2C, 2 × SO3CH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for C31H56NaO19S2 [M + Na]+: 819.2749; found: 819.2749.
Compound 60 was also obtained from the fully protected disaccharide 72 (200 mg, 0.213 mmol) by removal of the NAP-protecting group according to general method B. The crude product was purified by column chromatography (1:1 n-hexane/EtOAc) to give 60 (153 mg, 90%) as a colourless syrup.
Data of trisaccharide 61: Rf = 0.39 (n-hexane/EtOAc 4:6); [α]D24 = −20.0 (c = 0.12 in CHCl3); 1H NMR (400 MHz, CDCl3): δ = 5.50–5.48 (m, 2H, H-1′, H-1″), 4.67 (d, J1,2 = 3.3 Hz, 1H, H-1), 4.36–4.29 (m, 6H, 3 × SO3CH2CH3), 4.26 (t, J = 9.8 Hz, 1H, H-3), 4.09 (t, J = 9.7 Hz, 1H, H-3′), 3.83–3.64 (m, 7H, H-2, H-2′, H-2″, H-3″, H-5, H-5′, H-5″), 3.58–3.52 (m, 2H, H-4, H-4′), 3.43–3.41 (m, 1H, H-4), 3.40, 3.37, 3.36, 3.27, 3.24, 3.21, 3.20 (7 × s, 21H, C-1-OCH3, 6 × BDA-OCH3), 3.33–3.29 (m, 3H, 3 × H-7a), 3.18–3.11 (m, 3H, 3 × H-7b), 2.49 (s, 1H, H-4″-OH), 2.42–2.37 (m, 3H, 3 × H-6a), 2.06–1.86 (m, 3H, 3 × H-6b), 1.47–1.17 (m, 27H, 3 × SCH2CH3, 6 × BDA-CH3) ppm; 13C NMR (100 MHz, CDCl3): δ = 99.8, 99.7, 99.3, 99.2, 99.1 (6C, 6 × Cq BDA), 97.8 (1C, C-1), 97.6 (1C, C-1′), 97.1 (1C, C-1″), 76.5 (1C, C-4′), 76.2 (1C, C-4), 71.8 (1C, C-4″), 70.7 (1C, C-5″), 69.5, 69.3 (2C, C-3, C-3′), 68.8, 68.7, 68.4, 67.9, 66.6 (6C, C-2, C-2′, C-2″, C-3″, C-5, C-5′), 67.1, 67.0, 66.6 (3C, 3 × SO3CH2CH3), 55.3 (1C, C-1-OCH3), 48.2, 48.1, 48.0, 47.8, 47.6 (6C, 6 × BDA-OCH3), 46.7, 46.2 (3C, 3 × C-7), 27.4, 26.4 (3C, 3 × C-6), 17.9, 17.8, 17.4, 17.3 (6C, 6 × BDA-CH3), 15.3, 15.2, 15.1 (3C, 3 × SO3CH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C46H82O28S3 + Na]+: 1201.4047; found: 1201.4045.
Compound 61 was also obtained from the fully protected trisaccharide 74 (210 mg, 0.159 mmol) by removal of the NAP-protecting group according to general method B. The crude product was purified by column chromatography (4:6 n-hexane/EtOAc) to give 61 (137 mg, 73%) as a colourless syrup.
Methyl [2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-4-O-(2′-naphthyl)methyl-α-d-glucopyranosyl]-(1→4)-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (72).
Method I: To a solution of acceptor 59 (200 mg, 0.483 mmol) and donor 55 (457 mg, 0.724 mmol, 1.5 equiv.) in dry CH2Cl2 (10 mL), 4 Å molecular sieves (1.0 g) were added. The stirred mixture was cooled to −40 °C under argon. After 30 min at this temperature, a mixture of NIS (244 mg, 1.086 mmol, 1.5 equiv.) and TfOH (19 µL, 0.216 mmol) dissolved in THF (400 µL) was added and the reaction mixture was allowed to warm up to −15 °C in 1.5 h. The reaction mixture was diluted with CH2Cl2 (150 mL), washed successively with 10% aq. solution of Na2S2O3 (2 × 20 mL), satd. aq. solution of NaHCO3 (2 × 20 mL) and H2O (2 × 20 mL), dried over MgSO4 and concentrated. The crude product was purified by column chromatography (9:1 CH2Cl2/EtOAc) to give 72 (215 mg, 48%) as a colourless syrup.
Method II: To a solution of acceptor 59 (100 mg, 0.241 mmol) and donor 55 (213 mg, 0.338 mmol, 1.4 equiv.) in dry CH2Cl2 (5.0 mL), 4 Å molecular sieves (0.5 g) and sym-collidine (5.0 µL, 0.041 mmol, 0.12 equiv.) were added. The stirred mixture was cooled to −40 °C under argon. After 30 min at this temperature, a mixture of NIS (114 mg, 0.507 mmol, 1.5 equiv.) dissolved in dry THF (200 µL) and AgOTf (20 mg, 0.081 mmol) dissolved in dry toluene (250 µL) were added and the reaction mixture was allowed to warm up to 0 °C in 1.5 h. The reaction mixture was diluted with CH2Cl2 (100 mL), washed successively with 10% aq. solution of Na2S2O3 (2 × 15 mL), satd. aq. solution of NaHCO3 (2 × 15 mL) and H2O (2 × 15 mL), dried over MgSO4 and concentrated. The crude product was purified by column chromatography (9:1 CH2Cl2/EtOAc) to give 72 (158 mg, 70%) as a colourless syrup. Rf = 0.55 (CH2Cl2/EtOAc 9:1); [α]D24 = −33.3 (c = 0.09 in CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.83–7.44 (m, 7H, Ar-H), 5.57 (d, J1′,2′ = 4.2 Hz, 1H, H-1′), 5.10 (d, Jgem = 11.3 Hz, 1H, NpCH2a), 4.83 (d, Jgem = 11.3 Hz, 1H, NpCH2b), 4.66 (d, J1,2 = 3.4 Hz, 1H, H-1), 4.27 (t, J = 9.9 Hz, 1H, H-3), 4.22–4.16 (m, 4H, 2 × SO3CH2CH3), 4.08 (t, J = 9.1 Hz, 1H, H-3′), 3.82 (td, J = 2.2 Hz, J = 9.7 Hz, 1H, H-5), 3.77–3.67 (m, 3H, H-2, H-2′, H-5′), 3.57 (t, J = 9.3 Hz, 1H, H-4), 3.37, 3.36, 3.32, 3.25, 3.23 (5 × s, 15H, C-1-OCH3, 4 × BDA-OCH3), 3.35–3.34 (m, 1H, H-4′), 3.21–3.16 (m, 2H, H-7a, H-7a’), 3.10–3.04 (m, 2H, H-7b, H-7b’), 2.42–2.35 (m, 2H, H-6a, H-6a’), 2.03–2.00 (m, 1H, H-6b’), 1.88–1.85 (m, 1H, H-6b), 1.36–1.17 (m, 18H, 2 × SCH2CH3, 4 × BDA-CH3) ppm; 13C NMR (100 MHz, CDCl3): δ = 135.5, 133.2, 133.0 (3C, Cq Ar), 128.1–125.9 (7C, Ar), 99.8, 99.6, 99.3, 99.1 (4C, 4 × Cq BDA), 97.7 (1C, C-1), 96.6 (1C, C-1′), 78.1 (1C, C-4′), 75.3 (1C, C-4), 74.8 (1C, NpCH2), 70.2 (1C, C-3′), 69.7 (2C, C-3, C-5′), 68.3 (C-2′), 67.7 (2C, C-2, C-5), 66.7, 66.1 (2C, 2 × SO3CH2CH3), 55.1 (1C, C-1-OCH3), 48.0, 47.6 (4C, 4 × BDA-OCH3), 46.7 (1C, C-7), 46.4 (1C, C-7′), 27.2 (1C, C-6), 26.2 (1C, C-6′), 18.0, 17.8, 17.6, 17.3 (4C, 4 × BDA-CH3), 15.0, 14.9 (2C, 2 × SO3CH2CH3) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C42H64O19S2 + Na]+: 959.3375; found: 959.3372.
Methyl (2,3,4-tri-O-sulfate-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl-(1→4)-2,3-di-O-sulfate-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranoside heptasodium salt (73). To the solution of compound 53 (45 mg, 0.081 mmol) in dry DMF (2.0 mL) SO3·Et3N complex (367 mg, 2.025 mmol, 5 equiv./OH) was added and the reaction mixture was stirred at 50 °C for 24 h. The reaction was quenched with saturated aqueous solution of NaHCO3 (850 mg, 0.01 mmol), then, the solution was concentrated. The crude product was treated with Dowex ion-exchange resin (Na+ form) and purified by Sephadex G-25 column chromatography eluting with H2O to give 73 (68 mg, 78%) as a white solid. Rf = 0.35 (CH2Cl2/MeOH/H2O 6:7:1); [α]D24 = +8.7 (c = 0.15 in H2O); 1H NMR (400 MHz, D2O): δ = 5.44 (d, J1′,2′ = 4.7 Hz, 1H, H-1′), 5.04 (d, J1,2 = 5.7 Hz, 1H, H-1), 4.98 (t, J = 4.8 Hz, 1H, H-3′), 4.67–4.63 (m, 2H, H-2′, H-3), 4.36–4.33 (m, 2H, H-2, H-4′), 4.15–4.12 (m, 1H, H-5′), 3.86–3.82 (m, 1H, H-5), 3.76 (t, J = 9.9 Hz, 1H, H-4), 3.37 (s, 3H, C-1-O-CH3), 3.10–2.92 (m, 4H, H-7a,b, H-7‘a,b), 2.36–1.84 (m, 4H, H-6a,b, H-6′a,b) ppm; 13C NMR (100 MHz, D2O): δ = 96.7 (1C, C-1), 92.8 (1C, C-1′), 77.9 (1C, C-3), 76.1 (1C, C-4), 75.1 (2C, C-2, C-4′), 74.1 (1C, C-3′), 72.6 (1C, C-5′), 72.3 (1C, C-2′), 68.4 (1C, C-5), 55.1 (1C, C-1-OCH3), 47.2 (2C, C-7, C-7′), 26.6 (2C, C-6, C-6′) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C15H21Na7O30S7 + 2Na]2+: 555.8615; found: 555.8614.
Methyl [2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-4-O-(2′-naphthyl)methyl-α-
d-glucopyranosyl]-(1→4)-[2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-
d-glucopyranosyl]-(1→4)-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-6-deoxy-6-C-(ethylsulfonatomethyl)-α-
d-glucopyranoside (
74). To a solution of acceptor
60 (230 mg, 0.289 mmol) and donor
55 [
24] (274 mg, 0.433 mmol, 1.5 equiv.) in dry CH
2Cl
2 (5.0 mL), 4 Å molecular sieves (1.0 g) and
sym-collidine (7.0 µL, 0.052 mmol, 0.12 equiv.) were added. The stirred mixture was cooled to −40 °C under argon. After 30 min at this temperature, a mixture of NIS (146 mg, 0.649 mmol, 1.5 equiv.) dissolved in dry THF (200 µL) and AgOTf (27 mg, 0.104 mmol, 0.24 equiv.) dissolved in dry toluene (200 µL) was added and the reaction mixture was allowed to warm up to 10 °C in 1.5 h. The reaction mixture was diluted with CH
2Cl
2 (150 mL), washed successively with 10% aq. solution of Na
2S
2O
3 (2 × 20 mL), satd. aq. solution of NaHCO
3 (2 × 20 mL) and H
2O (2 × 20 mL), dried over MgSO
4 and concentrated. The crude product was purified by column chromatography (1:1
n-hexane/EtOAc) to give
74 (226 mg, 60%) as a colourless syrup.
Rf = 0.25 (
n-hexane/EtOAc 1:1); [α]
D24 = −14.0 (
c = 0.10 in CHCl
3);
1H NMR (400 MHz, CDCl
3):
δ = 7.83–7.43 (m, 7H, Ar-H), 5.50–5.48 (m, 2H, H-1′, H-1″), 5.10 (d,
Jgem = 11.3 Hz, 1H, NpC
H2a), 4.82 (d,
Jgem = 11.3 Hz, 1H, NpC
H2b), 4.66 (d,
J1,2 = 3.3 Hz, 1H, H-1), 4.28–4.02 (m, 8H, 3 × SO
3C
H2CH
3, H-3, H-3′), 3.80–3.70 (m, 6H, H-2, H-2′, H-2″, H-5, H-5′, H-5″), 3.57–3.51 (m, 3H, H-3″, H-4, H-4′), 3.38, 3.37, 3.27, 3.26, 3.23, 3.21 (6 × s, 21H, C-1-OC
H3, 6 × BDA-OC
H3), 3.36–3.34 (m, 1H, H-4), 3.33–3.31 (m, 3H, 3 × H-7a), 3.17–3.13 (m, 3H, 3 × H-7b), 2.41–2.38 (m, 3H, 3 × H-6a), 2.03–1.85 (m, 3H, 3 × H-6b), 1.44–1.17 (m, 27H, 3 × SCH
2C
H3, 6 × BDA-C
H3) ppm;
13C NMR (100 MHz, CDCl
3):
δ = 135.6, 133.2, 133.0 (3C, C
q Ar), 128.1–125.9 (7C, Ar), 99.8, 99.7, 99.2, 99.1, 99.0 (6C, 6 × C
q BDA), 97.7 (1C, C-1), 97.2 (1C, C-1′), 97.0 (1C, C-1″), 78.5 (1C, C-4″), 76.5 (1C, C-4′), 76.1 (1C, C-4), 74.9 (1C, Np
CH
2), 69.9, 69.5, 69.3, 68.7, 68.3, 67.8 (9C, C-2, C-2′, C-2″, C-3, C-3′, C-3″, C-5, C-5′, C-5″), 67.9, 66.6 (3C, 3 × SO
3CH
2CH
3), 55.2 (1C, C-1-O
CH
3), 48.2, 48.1, 48.0, 47.8, 47.6 (6C, 6 × BDA-O
CH
3), 46.6, 46.3, 46.0 (3C, 3 × C-7), 27.6, 27.4, 26.4 (3C, 3 × C-6), 17.9, 17.8, 17.7, 17.3, 17.2 (6C, 6 × BDA-
CH
3), 15.2, 15.0, 14.9 (3C, 3 × SO
3CH
2CH
3) ppm; UHR ESI-QTOF (positive ion):
m/
z calcd for [C
57H
90O
28S
3 + 2Na]
2+: 682.2283; found: 682.2292.
Methyl 2,3,4-tri-O-sulfate-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl-(1→4)-2,3-di-O-sulfate-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl-(1→4)-2,3-di-O-sulfate-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranoside decasodium salt (75). To the solution of compound 54 (20 mg, 0.024 mmol) in dry DMF (600 µL), SO3·Et3N complex (152 mg, 0.840 mmol, 5 equiv./OH) was added and the reaction mixture was stirred at 50 °C for 24 h. The reaction was quenched with saturated aqueous solution of NaHCO3 (850 mg, 0.01 mmol), then, the solution was concentrated. The crude product was treated with Dowex ion-exchange resin (Na+ form) and purified by Sephadex G-25 column chromatography eluting with H2O to give 75 (31 mg, 84%) as a white solid. Rf = 0.07 (CH2Cl2/MeOH/H2O 6:7:1); [α]D24 = +63.8 (c = 0.13 in H2O); 1H NMR (400 MHz, D2O): δ = 5.55 (s, 2H, H-1′, H-1″), 5.13 (d, J1,2 = 3.6 Hz, 1H, H-1), 4.99–4.97 (m, 1H, H-3′), 4.84–4.71 (m, 3H, H-2′, H-3, H-3″), 4.52 (dd, J = 8.6 Hz, J = 2.4 Hz, 1H, H-2″), 4.41 (dd, J = 9.7 Hz, J = 3.6 Hz, 1H, H-2), 4.31–4.24 (m, 2H, H-4″, H-5′), 3.99–3.91 (m, 3H, H-4′, H-5, H-5″), 3.84 (t, J = 9.0 Hz, 1H, H-4), 3.45 (s, 3H, C-1-O-CH3), 3.24–2.98 (m, 6H, H-7a,b, H-7‘a,b, H-7″a,b), 2.52–2.41 (m, 2H, H-6a, H-6″a), 2.28–2.18 (m, 2H, H-6′a,b), 2.11–1.90 (m, 2H, H-6b, H-6″b) ppm; 13C NMR (100 MHz, D2O): δ = 97.9 (1C, C-1), 94.9, 93.7 (2C, C-1′, C-1″), 79.2 (1C, C-3), 77.9 (1C, C-4′), 76.8 (2C, C-2, C-4), 76.3 (2C, C-3″, C-5″), 75.6 (1C, C-3′), 75.2 (1C, C-2′), 73.4 (1C, C-2″), 73.2 (1C, C-5′), 71.5 (1C, C-4″), 69.5 (1C, C-5), 56.2 (1C, C-1-OCH3), 48.3, 48.2, 48.0 (3C, C-7, C-7′, C-7″), 28.1, 27.7, 27.3 (3C, C-6, C-6′, C-6″) ppm; UHR ESI-QTOF (positive ion): m/z calcd for [C22H30Na10O43S10 + 2Na]2+: 788.8065; found: 788.8063.

 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}