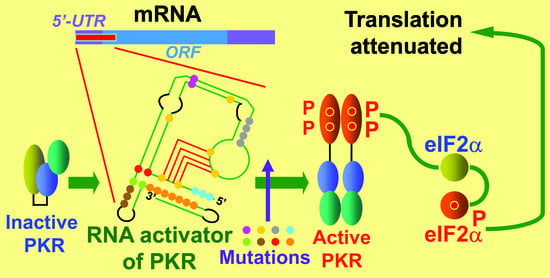
2.1. The RNA Activator of PKR within Human IFNG mRNA That Controls mRNA Translation
Before entering into the analysis of mutations within the
IFNG gene that may lead to RNA-mediated human disease, it is important to consider first the
IFNG regulatory circuit involving PKR activation.
IFNG provides the first example of a gene that controls the translation of its own mRNA by over an order of magnitude through the device of local activation of PKR by
IFNG mRNA [
15]. Indeed, overexpression of IFN-γ protein occurs in many autoimmune diseases [
18] as well as in toxic shock induced by bacterial superantigen toxins [
19,
20]. To avoid a harmful hyper-inflammatory response, the
IFNG gene expresses within its mRNA a 203-nucleotide RNA pseudoknot element located within the 5′-untranslated region (UTR) and the start of the open reading frame (
Figure 1) that potently activates PKR locally in
cis [
15,
16]. This in turn leads to a local phosphorylation of eIF2α within the vicinity of
IFNG mRNA that strongly diminishes the rate of initiation of translation of this mRNA while leaving the translation of other mRNA species in the cell intact. Dynamic refolding of the RNA pseudoknot enables
IFNG mRNA to function both as a potent activator of PKR and as an RNA template for translation [
16]. This autoregulatory circuit renders the translation of
IFNG mRNA sensitive to viral inhibitors of PKR such as Vaccinia virus E3L, leading to significantly higher expression of IFN-γ upon viral infection [
15]. PKR is expressed constitutively in the cell [
21] to levels that support this
IFNG autoregulatory mechanism [
15].
The
a4 mutation (U9G A14C A29G A45U; yellow in
Figure 1) severely impairs the activation of PKR. Restoration of A14 in the
a4 mutant sufficed to restore the ability to activate PKR. The
d1 deletion of the 5′-terminal nucleotides through U9 (cyan, light green, and yellow in
Figure 1) leads to nearly complete loss of activation of PKR and increases the translation efficiency of wild type
IFNG mRNA by more than tenfold. Mutations U9 and A14 each affect the stability of the pseudoknot stem (red text in
Figure 1). In PKR knockout cells, translation efficiency of wild type (
wt)
IFNG mRNA was increased to that of
a4 and
d1 mRNA. The
a4r mutation (U64G A69C; yellow in
Figure 1) likewise enhanced translation efficiency of
IFNG mRNA, by 7-fold, whereas the compensatory
a4/a4r mutation restored the stability of the pseudoknot stem and the activation of PKR and lowered the translation efficiency to almost that of
wt mRNA. These results render mutation of either nucleotide U9, A14, U64, or A69 within the 5′-UTR of the
IFNG gene critical targets for inflammatory disease, leading to excessive expression of IFN-γ protein by around an order of magnitude [
15].
Moreover, the U7A mutation (light green in
Figure 1) abrogates PKR activation and enhances mRNA translation by 6-fold, showing that U7 is critical for proper function [
16].
At the junction of helices
S1 and
S2, the A120:U15 base pair (red in
Figure 1) is critical for PKR activation. Mutation of this pair to G:C resulted in near-total loss of the ability to activate PKR. Moreover, mere inversion of the A120:U15 pair sufficed to abolish PKR activation. By contrast, when all of the nucleotides in helix
S1 (U15-A29:U103-A120) except the A120:U15 pair were replaced by a theophyllin aptamer and slippery helix [
22], the ability to activate PKR remained intact [
16]. These results revealed that the orientation of the A120:U15 pair located at the helical junction in
S1 is critical for PKR activation.
Helix
S2 is likewise critical for PKR activation. Replacement of nucleotides 121–127 (green in
Figure 1), including the AUG start codon, by the nucleotides in the opposite strand abolished the ability to activate PKR (mutation
s2a). By contrast, replacement of nucleotides 163–169 by the nucleotides in the opposite strand left the ability to activate PKR intact (mutation
s2b). Restoration of base pairing by an inverted S2 helix (
s2ab) failed to restore the ability to activate PKR. However, replacement of the helical junction base pair U121:A169 within
s2ab by the original A121:U169 (light green in
Figure 1) led to full recovery of the ability to activate PKR, demonstrating that A121-U169 is a critical, orientation-sensitive base pair for PKR activation. Replacement of the bifurcation loop adjoining the AUG start codon (nucleotides 128–162) by pentaloop UUCGU left the ability to activate PKR largely intact, showing that the bifurcation loop is not critical for this property. The A121-G127 strand in helix
S2 can pair alternatively with the U7-C1 5′-terminal strand for PKR activation. Like the A121:U169 base pair, the A121:U7 pair is orientation-sensitive, as seen from the U7A mutation reported above, which abrogates PKR activation. Even when
S2 is intact, deletion of the first six nucleotides C1-U6 (cyan in
Figure 1) results in marked loss of the ability to activate PKR, and to complete loss when U7 is also deleted [
16].
A single nucleotide change within helix
S3, A202U (light blue in
Figure 1), sufficed to abolish PKR activation. By contrast, the U171A single mutation did retain partial ability to activate PKR. However, U171A could not rescue A202U, as the U171A:A202U double mutation completely lost the ability to activate PKR. This revealed that as for A120:U15, the U171:A202 base pair is orientation-sensitive for PKR activation. Hence, A120 and A202 are potential mutation targets for inflammatory disease. The remainder of helix
S3 is also sensitive to mutation;
s3a (orange in
Figure 1), in which U171-C184 are replaced by the nucleotides in the opposite strand, abrogated the ability to activate PKR [
16].
Moreover, U170 adjoining the
S2 helix is located in a sensitive position; mutation U170A (brown in
Figure 1) resulted in loss of PKR activation [
16].
Formation of a kink-turn (K-turn) is known to depend on purine–purine pairing; a G•A/A•G quartet underlies most K-turns in prokaryotic RNA [
23]. Indeed, in the
IFNG K-turn, mutation G38C (purple,
Figure 1) severely reduced the activation of PKR and led to an enhancement of translation efficiency even greater than that caused by the
d1 mutation [
15,
16]. Moreover, mutation A95G (purple in
Figure 1) similarly impaired the ability to activate PKR, pointing to a G•A/A•A motif as essential for PKR activation by
IFNG mRNA. Typical K-turns have G:C base pairs bordering the bulge [
23], yet neither U92C nor G36C mutation (light purple,
Figure 1) affected the activation of PKR [
16].
Within the loop bordering the pseudoknot stem U64-G70, no detrimental mutations could be detected. However, replacement of the four nucleotides 78CAAG81 (grey in
Figure 1) in the short stem by those of the opposite strand (78GUUU81) resulted in near-total loss of the ability to activate PKR, showing that this short stem is critical for function [
16].
Thus, throughout its 203-nucleotide sequence, the IFNG RNA element that activates PKR is extremely sensitive to single nucleotide mutations, rendering it a prime target for human inflammatory disease.
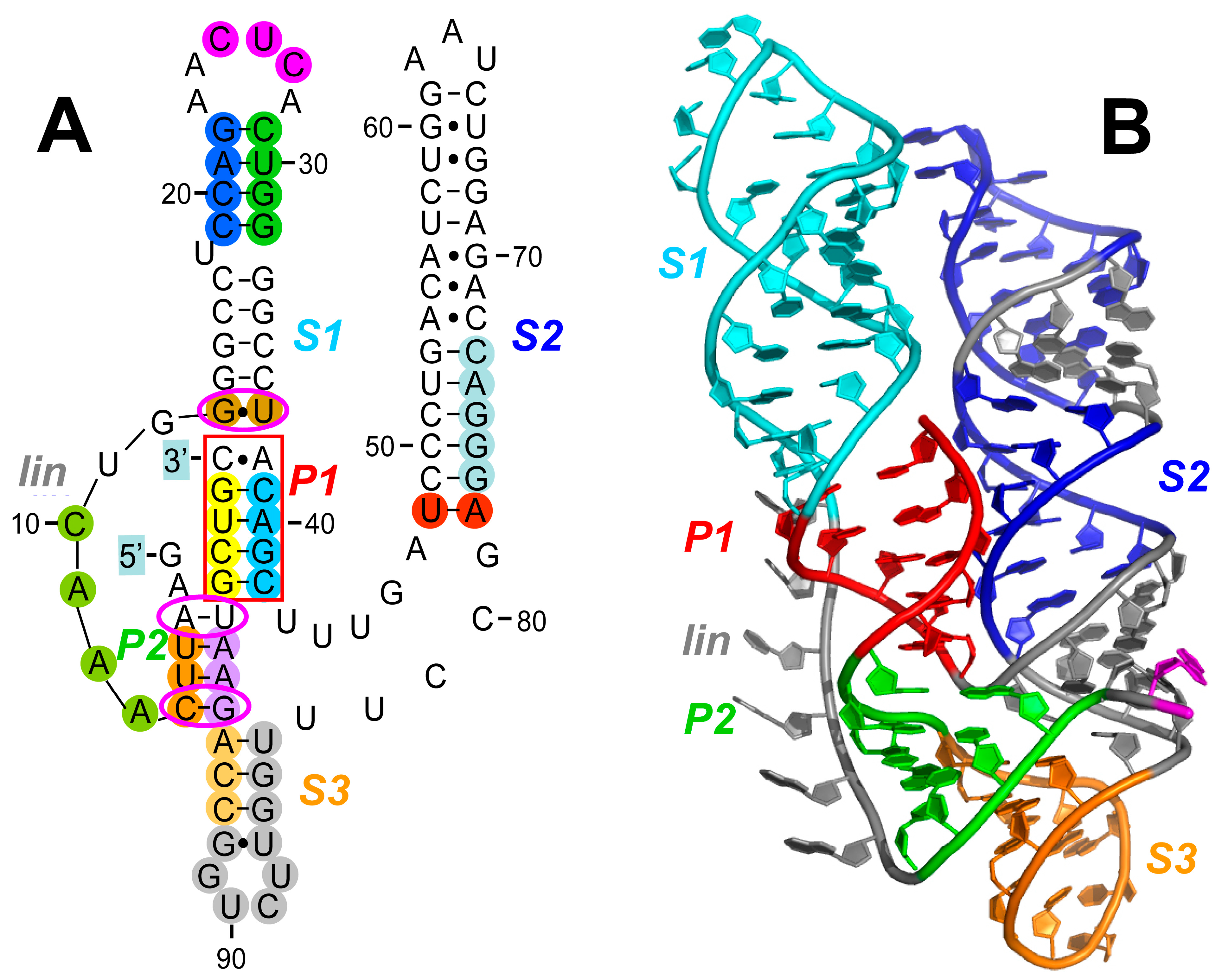
2.2. The RNA Activator of PKR within Human TNF Pre-mRNA That Controls mRNA Splicing
TNFα (
TNF) provides the first example of a gene that regulates splicing of its own pre-mRNA by over an order of magnitude through the device of local activation of PKR within the nucleus by its pre-mRNA, leading to the phosphorylation of eIF2α that enables efficient splicing [
13,
14,
24]. The 104-nucleotide RNA activator of PKR within the human
TNF gene was designated as the 2-aminopurine response element (2-APRE) [
13] because 2-aminopurine, a small-molecule blocker of PKR activation and action, inhibits the splicing of
TNF pre-mRNA [
13,
24]. The TNFα cytokine is produced early during an immune response. Upon immune stimulation,
TNFα mRNA is expressed rapidly, reaching its maximum within 3 h [
24], whereas the highly homologous
TNFβ (
lymphotoxin) mRNA is still increasing by 24 h. The difference is vested in the
TNFα 2-APRE, because insertion of this element into the
TNFβ 3′-UTR sufficed to increase
TNFβ RNA splicing efficiency by an order of magnitude [
13,
14]. Increased expression of PKR within the cell causes a strong increase in splicing of
TNF mRNA [
13]. The 2-APRE RNA sequence of 104 nucleotides folds into a compact pseudoknot (
Figure 2). This pseudoknot constrains the RNA into two double-helical stacks with parallel axes (
Figure 2B) that are each of sufficient length to bind a PKR monomer, enabling PKR dimerization that is needed for its local activation [
14]. Within the nucleus, phosphorylation of eIF2α substrate upon PKR activation is not only strictly needed for splicing of
TNF pre-mRNA but is even sufficient to promote highly efficient splicing [
14]. eIF2α phosphorylation upregulates
TNF mRNA splicing in human peripheral blood mononuclear cells, showing its physiological relevance. Once exported from the nucleus,
TNF mRNA is translated without inhibition by PKR activation [
13,
14], presumably because cytoplasmic proteins shield the 2-APRE [
13].
Long stem–loop
S1 forms an integral part of the
TNF RNA activator of PKR (cyan in
Figure 2). Helix
S1 forms a domain that is sensitive to mutation. Mutating 19CCAG22 to the four nucleotides in the complementary strand 19GGUC22, yielding mutation
S1a (blue in
Figure 2A), reduced the ability to activate PKR by more than twofold. Mutating in addition 29CUGG32 to 29GACC32 to create double mutation
S1ab (blue and green in
Figure 2A) only slightly improved PKR activation. The loop of helix
S1 in the 2-APRE is also sensitive to mutation. Thus, mutating nucleotides 25CUC27 to 25GCG27, yielding mutation
S1L (purple in
Figure 2A), reduced the ability to activate PKR to the same extent as mutation
S1a.
The
TNF 2-APRE pseudoknot is created around two pseudoknot stems,
P1 (red in
Figure 2) and
P2 (green in
Figure 2). Pseudoknot stem
P1 (red box in
Figure 2A) is critical for PKR activation. Mutation of 39CAGC42 to the nucleotides in the opposite strand, 39GUCG42 (
P1a mutation, light blue in
Figure 2A), severely impaired PKR activation, as did mutation of 100GCUG103 to 100CGAC103 (
P1b mutation, yellow in
Figure 2A). Activation of PKR was restored in part by the
P1ab double mutation.
Pseudoknot stem
P2 is required for full PKR activation. Mutation of 4UUC6 to the nucleotides in the opposite strand, 4AAG6 (
P2b mutation, orange in
Figure 2A), somewhat impaired PKR activation. Activation of PKR was restored largely by also mutating 96GAA98 to 96CUU98 (
P2a mutation, light purple in
Figure 2A), creating the
P2ab double mutation.
The indispensable role of helix
S3 (orange in
Figure 2) bordering
P2 is highlighted by the
S3bP2b double mutation which changes 93CCAGAA98 into 93GGUCUU98 (pale orange and light purple in
Figure 2A), to thereby abrogate the ability to activate PKR.
The
TNF RNA activator of PKR relies tightly on the base pairs adjoining pseudoknot stem
P1 and at the terminus of stem
P2 (three purple ellipses in
Figure 2A). Thus, mutations G13C and G13A each weakened the ability to activate PKR, whereas mutation of U37C largely abrogated PKR activation. This shows that the 13G•U37 wobble base pair (light brown in
Figure 2A) at the start of helix
S1 is highly specific for function; neither 13C•U37 nor 13G•C37 can replace it. Each of the A3U and U99A mutations strongly impaired PKR activation, which could not be restored in the A3U U99A double mutation, showing that the 3A:U99 base pair is orientation-sensitive. By contrast, each of the G96C and C6G mutations strongly impaired PKR activation, which could be restored largely by the G96C C6G double mutation.
The long stem–loop
S2 (dark blue in
Figure 2B) forms an integral part of the
TNF RNA activator of PKR. The U48•A78 base pair at the base of long helix
S2 in the 2-APRE (red in
Figure 2A) is critical for the ability to activate PKR. Mutations U48A and A78U each severely reduce the activation of PKR by 2-APRE RNA, as does double mutation U48A A78U, showing that the U48•A78 pair is orientation-sensitive. By contrast, mutation of 49CCCUG53 in helix
S2 to 49GGGAA53 (S2aUA mutation) left activation of PKR intact. By contrast,
S2bUA mutation of 73CAGGG77 in helix
S2 to 73UUCCC77 (light blue in
Figure 2A) impaired PKR activation. However, double mutation
S2abUA impaired PKR activation only weakly, showing that this part of helix
S2 adjacent to the U48•A78 pair is more tolerant to mutation.
The importance of stem–loop
S3 nucleotides (pale orange and grey in
Figure 2A; orange in
Figure 2B), which joins the two parallel helix motifs
S1-P1-P2 and
S2 within the folded pseudoknot structure (
Figure 2B), is documented by the deletion of nucleotides 84–95 (grey and orange in
Figure 2A), which weakens the ability to activate PKR.
The six-nucleotide linker (
lin) (grey in
Figure 2), which connects pseudoknot stem
P2 to helix
S1, is highly sensitive to deletion, as might be expected. Indeed, deleting nucleotides 7AAAC10 (light green in
Figure 2A) totally abolishes the ability to activate PKR. Moreover, mutating 5UCAAA9 to the complementary nucleotides 5AGUUU9 while maintaining the linker length, to generate the
P2 lin(5–9) mutation (orange and light green in
Figure 2A), strongly impaired the ability to activate PKR. In this context, 5UC6 (orange in
Figure 2A), which form part of pseudoknot stem P2, are the two critical nucleotides, because mutation of 7AAA9 alone to 7UUU9 failed to impair PKR activation.
In conclusion, throughout its 104-nucleotide sequence, the TNF RNA element that activates PKR is most sensitive to mutations, rendering it, like the IFNG RNA activator of PKR, a prime target for inflammatory disease upon mutation.
2.3. The RNA Activator of PKR and Silencer of PKR Activation within Human Globin Pre-mRNA That Controls mRNA Splicing
Efficient splicing of adult
α-globin and
β-globin as well as of fetal
Aγ-globin pre-mRNA species requires activation of PKR and eIF2α phosphorylation within the nucleus [
17]. Splicing of
β-globin mRNA in intact cells is abrogated by 2-aminopurine or by co-expression of dominant-negative mutant PKR [
17]. Like
IFN-γ mRNA,
β-globin pre-mRNA activates PKR in vitro [
17]. Once activated, PKR must phosphorylate its eIF-2α substrate to promote assembly of early spliceosomes and to enable efficient splicing of
β-globin and
α-globin mRNA, a key step towards erythropoiesis. Expression of non-phosphorylatable mutant eIF2αS51A inhibits splicing in cells, as do antibodies against phospho-eIF2α in vitro [
17]. Once
β-globin intron 1 is excised, sequence
S1c near the 5′-terminus of exon 2 (red in
Figure 3) induces a structural rearrangement within the RNA that silences the ability of spliced
β-globin mRNA to activate PKR. As a result, activation of PKR is highly transient, serving only to enhance splicing while avoiding inhibition of β-globin protein synthesis [
17,
25].
The 5-terminal 124 nucleotides in
β-globin exon 1, comprising the 50-nucleotide 5′-UTR and the first 74 nucleotides of the open reading frame, constitute the RNA activator of PKR (
Figure 3A). Deletion of nucleotides 119AGUUGG124 (purple in
Figure 3A) led to almost complete loss of the ability to activate PKR; deletion of nucleotides 1ACAU4 from the 5′-end (light purple in
Figure 3A) strongly reduced PKR activation.
Mutation
s1a of 47CACCA51 (yellow in
Figure 3A) to the nucleotides in the opposite strand reduced the ability to activate PKR, whereas mutation
s1b of 110CGUGG114 (grey in
Figure 3A) to the nucleotides in the opposite strand completely abrogated the ability to activate PKR. Combining both mutations, in
s1ab, created a new 5-base pair helix and fully restored the activation of PKR, showing that this part of the
β-globin RNA activator of PKR is not orientation-sensitive. Hence, the
S1a-S1b helix in
Figure 3A constitutes an essential core of the
β-globin RNA activator of PKR, rendering it a sensitive target for mutational disease.
During the differentiation of erythroid cells, there is a dramatic increase in the cellular globin content, which increases from less than 0.1% of total protein in the proerythroblast to 95% in the reticulocyte, reflecting a massive translation of globin mRNA [
26]. Erythropoiesis would be compromised severely if the mature spliced
β-globin mRNA should retain the ability to activate PKR and thereby inhibit its own translation severely, as exemplified by
IFNG mRNA [
15] reviewed above. This defines a need for a mechanism that silences the ability of mature
β-globin mRNA to activate PKR once splicing has taken place.
Once intron 1 is excised, and exon 2 is joined to exon 1, the 5-nucleotide
S1c sequence U148-G152 (
Figure 3A) located within the first ten nucleotides of exon 2 in spliced
β-globin mRNA displaces strand
S1b from
S1a, which results in a major structural rearrangement (
Figure 3B) that silences the ability to activate PKR, thereby allowing unimpeded translation of
β-globin mRNA in the cytoplasm, essential for survival. Mutation of the 5′-CACCA-3′ motif in
S1a (yellow in
Figure 3B) to 5′-GUGGU-3′ restored the ability of the spliced
β-globin mRNA to activate PKR, as did mutation of the
S1c motif to 5′-ACCAC-3′. The double mutation, which restores base pairing between
S1a and
S1c, restored silencing of PKR activation even though the orientation of the two complementary strands was inverted [
17]. Accordingly, mutations within each of
S1a or
S1c are deleterious, rendering them highly sensitive targets for disease.
α-Globin pre-mRNA activates PKR as strongly as does
β-globin mRNA. Splicing of
α-globin pre-mRNA depends likewise on the activation of PKR and on eIF2α phosphorylation. However, within
α-globin pre-mRNA, the locations of PKR activator and silencer are reversed, the silencer mapping into exon 1 and the PKR activator into exon 2 [
17].
The two fetal
Aγ- and
Gγ-
globin genes are related more closely to the adult
β-globin gene. Except for a single nucleotide, at position 25 in the 5′-UTR,
Gγ- and
Aγ-
globin gene sequences are identical through exon 2 [
27]. Activation of PKR and phosphorylation of eIF2α also regulate excision of
Aγ-
globin intron 1 [
17]. The
Aγ-
globin RNA activator of PKR and the silencer element reside within exon 1 and the first 172 nucleotides of exon 2 [
17], but their precise locations have not yet been determined.
Hence, the β-globin gene, and by inference the α-globin and fetal γ-globin genes, each represent RNA targets for disease resulting from mutations that affect either their ability to activate PKR or their ability to silence PKR activation after mRNA splicing has taken place, which is critical for allowing the maximal rate of translation of the mature globin mRNA products, essential for hemoglobin production, for breathing and thus for the survival of humans.
The human gene mutation database for the
β-globin gene (HBB in
www.hgmd.org) contains a number of β-thalassemia mutations that map into the RNA activator of PKR or into the silencer element [
25]. These β-thalassemia mutations include 110C and 117G within the
S1b strand at the core of the PKR activator structure and U148 in the spliced mRNA that falls within the silencer sequence (
Figure 3) [
25]. However, it is not known as yet whether any of these numerous β-thalassemia mutations affect the ability of
β-globin pre-mRNA to activate PKR essential for maximal splicing, or to silence PKR activation after mRNA splicing has taken place, which is critical for the translation efficiency of
β-globin mRNA.




{kind=link}
{kind=link}
{kind=link}
{kind=link}


