4.2. Chemistry
4.2.1. General Procedure for Alpha-Benzamide Cyclobutanones (3a-3j, and 3s-3u)
To a solution of substituted benzoic acid (1 eq, 0.249 mmol) and acetal (±)-2 (1.2 eq, 0.298 mmol) in ethyl acetate (1.25 mL), N-methyl morpholine (5.0 eq, 1.24 mmol) was added. Then, propylphosphonic anhydride (2.5 eq, 0.62 mmol) was added to the above mixture as a solution in ethyl acetate (purchased as ≥50 weight % in EA). The reaction mixture was stirred under N2 at 80 °C for 48 h or until complete consumption of the benzoic acid determined with TLC (EA/hexane = 50/50 and ~3 to 4 drops of glacial acetic). The reaction was quenched by adding water (3 mL); then, the organic product was extracted using ethyl acetate (3 × 3 mL). The combined organic layers were washed successively with water (3 × 3 mL) and 1N HCl (3 mL) and then dried over Na2SO4. The solvent was removed via evaporation under reduced pressure providing the corresponding acetal intermediate. Without further purification, crude acetal (1 eq) was subjected to hydrolysis conditions, wherein the crude mixture was dissolved in acetone (1.2 mL), water (0.17 mL, 10% v/v), and 1N HCl (0.34 mL, 30% v/v). Then, the reaction mixture was stirred at 50 °C for 18 h with periodic TLC and HPLC monitoring. Upon completion, the organic product was extracted using ethyl acetate (3 × 3 mL) or methylene chloride for highly water-soluble analogs, and the combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure, and the crude mixture was purified via column chromatography to afford the corresponding alpha-benzamide cyclobutanone.
2-Methyl-N-(2-oxocyclobutyl)benzamide (3a). The crude product of 3a was purified via column chromatography using ethyl acetate/hexane (40/60) to afford compound 3a as a white solid (35.7 mg, 71%): mp 100–102 °C. 1H NMR (500 MHz, CDCl3) δ 7.40 (dd, J = 7.6, 1.5 Hz, 1H), 7.35 (td, J = 7.5, 1.5 Hz, 1H), 7.30–7.18 (m, 2H), 6.30 (NH, d, J = 7.8 Hz, 1H), 5.21–5.12 (m, 1H), 3.10–2.96 (m, 2H), 2.63–2.52 (m, 1H), 2.47 (s, 3H), 2.16 (dtd, J = 11.0, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.0, 169.4, 136.6, 134.8, 131.2, 130.4, 126.8, 125.8, 64.5, 42.3, 19.9. HRMS (ESI) calcd for M+Na+ C12H13NO2: 226.0838, found (M+Na+) 226.0840.
3-Methyl-N-(2-oxocyclobutyl)benzamide (3b). The crude product of 3b was purified via column chromatography on a Teledyne Isco Rf Flash chromatography unit (Teledyne ISCO, Lincoln, NE, USA) eluting with ethyl acetate/hexane (20/80) to afford compound 3b as a white crystalline solid (25.5 mg, 50%): mp 94–96 °C. 1H NMR (500 MHz, CDCl3) δ 7.52 (s, 1H), 7.51–7.43 (m, 1H), 7.32–7.21 (m, 2H), 6.56 (NH, d, J = 7.4 Hz, 1H), 5.14–5.05 (m, 1H), 2.95 (dd, J = 9.5, 7.8 Hz, 2H), 2.53–2.43 (m, 1H), 2.32 (d, J = 0.8 Hz, 3H), 2.07 (dtd, J = 11.0, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 204.1, 166.0, 137.5, 132.1, 131.7, 127.6, 127.5, 126.8, 123.0, 76.3, 76.0, 75.7, 63.6, 41.2, 20.3, 18.9. HRMS (ESI) calcd for M+Na+ C12H13NO2: 226.0838, found (M+Na+) 226.083.
4-Methyl-N-(2-oxocyclobutyl)benzamide (3c). The crude product of 3c was purified via column chromatography using ethyl acetate/hexane (40/60) to afford compound 3c as a white solid (27.4 mg, 50.6%): mp 149–150 °C. 1H NMR (500 MHz, CDCl3) δ 7.68 (d, 2H), 7.23 (d, J = 7.9 Hz, 2H), 6.80 (NH, d, J = 7.6 Hz, 1H), 5.19–5.10 (m, 1H), 3.05–2.96 (m, 2H), 2.59–2.48 (m, 1H), 2.40 (s, 3H), 2.18 (dtd, J = 11.0, 9.6, 8.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.6, 166.8, 142.5, 130.3, 129.3, 127.1, 64.6, 42.2, 21.5, 19.8. HRMS (ESI) calcd for M+Na+ C12H13NO2: 226.0838, found (M+Na+) 226.0840.
2-Chloro-N-(2-oxocyclobutyl)benzamide (3d). The crude product of 3d was purified via column chromatography on a Teledyne Isco Rf Flash chromatography unit eluting with ethyl acetate/hexane (40/60) to afford cyclobutanone 3d as a white crystalline solid (32.5 mg, 59%): mp 102–103 °C. 1H NMR (500 MHz, CDCl3) δ 7.71 (dd, J = 7.6, 1.7 Hz, 1H), 7.45–7.35 (m, 2H), 7.33 (td, J = 7.3, 1.9 Hz, 1H), 6.81 (NH, d, J = 7.2 Hz, 1H), 5.16 (dt, J = 10.3, 7.9 Hz, 1H), 3.03 (dd, J = 9.6, 7.7 Hz, 2H), 2.57 (tt, J = 10.6, 7.2 Hz, 1H), 2.24–2.13 (m, 1H). 1H NMR (500 MHz, CDCl3) δ 7.76–7.70 (m, 1H), 7.46–7.35 (m, 2H), 7.35 (ddd, J = 7.5, 6.8, 1.8 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 5.23–5.14 (m, 1H), 3.10–3.01 (m, 2H), 2.64–2.53 (m, 1H), 2.20 (dtd, J = 11.0, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 204.4, 165.8, 133.5, 131.9, 130.8, 130.6, 130.4, 127.2, 64.5, 42.4, 19.7. HRMS (ESI) calcd for MNa+ C11H10ClNO2: 246.0291, found (M+Na+) 246.0292.
3-Chloro-N-(2-oxocyclobutyl)benzamide (3e). The crude product of 3e was purified via column chromatography using ethyl acetate/hexane (50/50) to afford compound 3e as a white solid (47.6 mg, 79%): mp 64–66 °C. 1H NMR (500 MHz, CDCl3) δ 7.77 (t, J = 1.9 Hz, 1H), 7.65 (dt, J = 7.7, 1.4 Hz, 1H), 7.50 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H), 6.82 (d, J = 7.1 Hz, 1H), 5.23–5.14 (m, 1H), 3.12–2.97 (m, 2H), 2.62–2.51 (m, 1H), 2.18 (dtd, J = 11.0, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.12, 165.61, 134.87, 132.04, 129.99, 127.52, 125.19, 64.51, 42.30, 19.74. HRMS (ESI) calcd for MNa+ C11H10ClNO2: 246.0292, found (M+Na+) 246.0280.
4-Bromo-N-(2-oxocyclobutyl)benzamide (3f). The crude product of 3f was purified via column chromatography on a Teledyne Isco Rf Flash chromatography unit eluting with ethyl acetate/hexane (30/70) to afford compound 3f as a white crystalline solid (23.7 mg, 36%): mp 104–105 °C. 1H NMR (500 MHz, CDCl3) δ 7.69–7.61 (m, 2H), 7.58 (dd, J = 8.6, 2.0 Hz, 2H), 6.85–6.77 (NH, s, 1H), 5.15 (tdd, J = 9.6, 7.8, 1.4 Hz, 1H), 3.04 (ddd, J = 9.5, 7.8, 1.6 Hz, 2H), 2.55 (dtdd, J = 12.3, 9.7, 6.9, 2.1 Hz, 1H), 2.19 (dtd, J = 11.0, 9.6, 8.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.2, 165.9, 131.9, 128.7, 126.8, 64.5, 42.3, 19.7.
3-Chloro-4-methoxy-N-(2-oxocyclobutyl)benzamide (3g). The crude product of 3g was purified via column chromatography on a Teledyne Isco Rf Flash chromatography unit eluting with ethyl acetate/hexane (40/60) to afford compound 3g as a white solid (29.5 mg, 47%): mp 134–136 °C. 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 2.2 Hz, 1H), 7.70–7.65 (m, 1H), 6.94 (d, J = 8.8 Hz, 1H), 6.63 (d, J = 7.4 Hz, 1H), 5.18–5.09 (m, 1H), 3.95 (s, 3H), 3.06–2.99 (m, 2H), 2.54 (tt, J = 10.5, 7.2 Hz, 1H), 2.21–2.10 (m, 1H). 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 2.3 Hz, 1H), 7.66 (dd, J = 8.6, 2.3 Hz, 1H), 6.97 (d, J = 7.7 Hz, 1H), 6.92 (d, J = 8.6 Hz, 1H), 5.12 (dt, J = 9.8, 7.6 Hz, 1H), 3.94 (s, 3H), 3.01 (dd, J = 9.4, 7.8 Hz, 2H), 2.57–2.46 (m, 1H), 2.20 (dtd, J = 11.0, 9.5, 8.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 206.0, 165.4, 157.8, 129.4, 127.3, 126.1, 122.7, 111.5, 64.5, 56.3, 42.2, 19.6.
2-Chloro-4,5-dimethoxy-N-(2-oxocyclobutyl)benzamide (3h). The crude product of 3h was purified via column chromatography eluting with ethyl acetate/hexane (60/40) to afford 3h as a white solid (38.2 mg, 54%): mp 123–125 °C. 1H NMR (500 MHz, CDCl3) δ 7.43 (s, 1H), 7.40–7.29 (m, 1H), 7.24 (NH, d, J = 7.5 Hz, 1H), 6.83 (s, 1H), 5.13–5.04 (m, 1H), 3.90 (d, J = 4.6 Hz, 6H), 3.14–2.85 (m, 2H), 2.59–2.42 (m, 1H), 2.24 (dddd, J = 11.0, 10.1, 9.0, 8.0 Hz, 1H). 1H NMR (500 MHz, CDCl3) δ 7.46 (s, 1H), 7.25 (d, J = 7.6 Hz, 1H), 6.85 (s, 1H), 5.10 (tdd, J = 10.0, 7.1, 2.2 Hz, 1H), 3.92 (d, J = 4.4 Hz, 6H), 3.15–3.05 (m, 1H), 3.08–2.98 (m, 1H), 2.55 (dtd, J = 11.0, 10.1, 5.1 Hz, 1H), 2.26 (dddd, J = 11.1, 10.1, 9.1, 8.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 204.8, 165.0, 151.5, 148.0, 124.2, 122.8, 113.6, 112.8, 64.8, 56.3, 56.2, 42.3, 19.6.
2-Hydroxy-N-(2-oxocyclobutyl)benzamide (3i). The crude mixture of 3i was purified via column chromatography using ethyl acetate/hexane (50/50) to afford cyclobutanone 3i as a white crystalline solid (49.0 mg, 88%): mp 149–151 °C. 1H NMR (500 MHz, CDCl3) δ 11.76 (OH, s, 1H), 7.40–7.28 (m, 1H), 7.28 (dd, J = 8.0, 1.6 Hz, 1H), 6.91 (dd, J = 8.4, 1.2 Hz, 1H), 6.88–6.73 (m, 2H), 5.10–5.01 (m, 1H), 3.05–2.92 (m, 2H), 2.55–2.43 (m, 1H), 2.12 (dtd, J = 11.1, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 203.5, 168.6, 160.7, 133.8, 124.6, 117.8, 117.7, 112.3, 62.9, 41.4, 18.7. HRMS (ESI) calcd for M+Na+ C11H12NO3: 228.0631, found (M+Na+) 228.0600.
2-Hydroxy-4-methoxy-N-(2-oxocyclobutyl)benzamide (3j). The crude product of 3j was purified via column chromatography using ethyl acetate/hexane (50/50) to afford cyclobutanone 3j as a white crystalline solid (38.4 mg, 88%): mp 131–133 °C. 1H NMR (500 MHz, CDCl3) δ 12.20 (s, 1H), 7.30–7.25 (m, 1H), 6.96 (NH, d, J = 7.7 Hz, 1H), 6.41 (d, J = 2.6 Hz, 1H), 6.37 (dd, J = 8.8, 2.6 Hz, 1H), 5.09 (dt, J = 9.9, 7.7 Hz, 1H), 3.79 (s, 3H), 3.01 (dd, J = 9.4, 7.8 Hz, 2H), 2.55–2.44 (m, 1H), 2.26–2.14 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 205.0, 169.5, 164.8, 164.0, 126.9, 107.3, 106.3, 101.6, 64.0, 55.5, 42.3, 19.8. HRMS (ESI) calcd for MNa+ C12H13NO4: 258.0737, found (M+Na+) 258.0730.
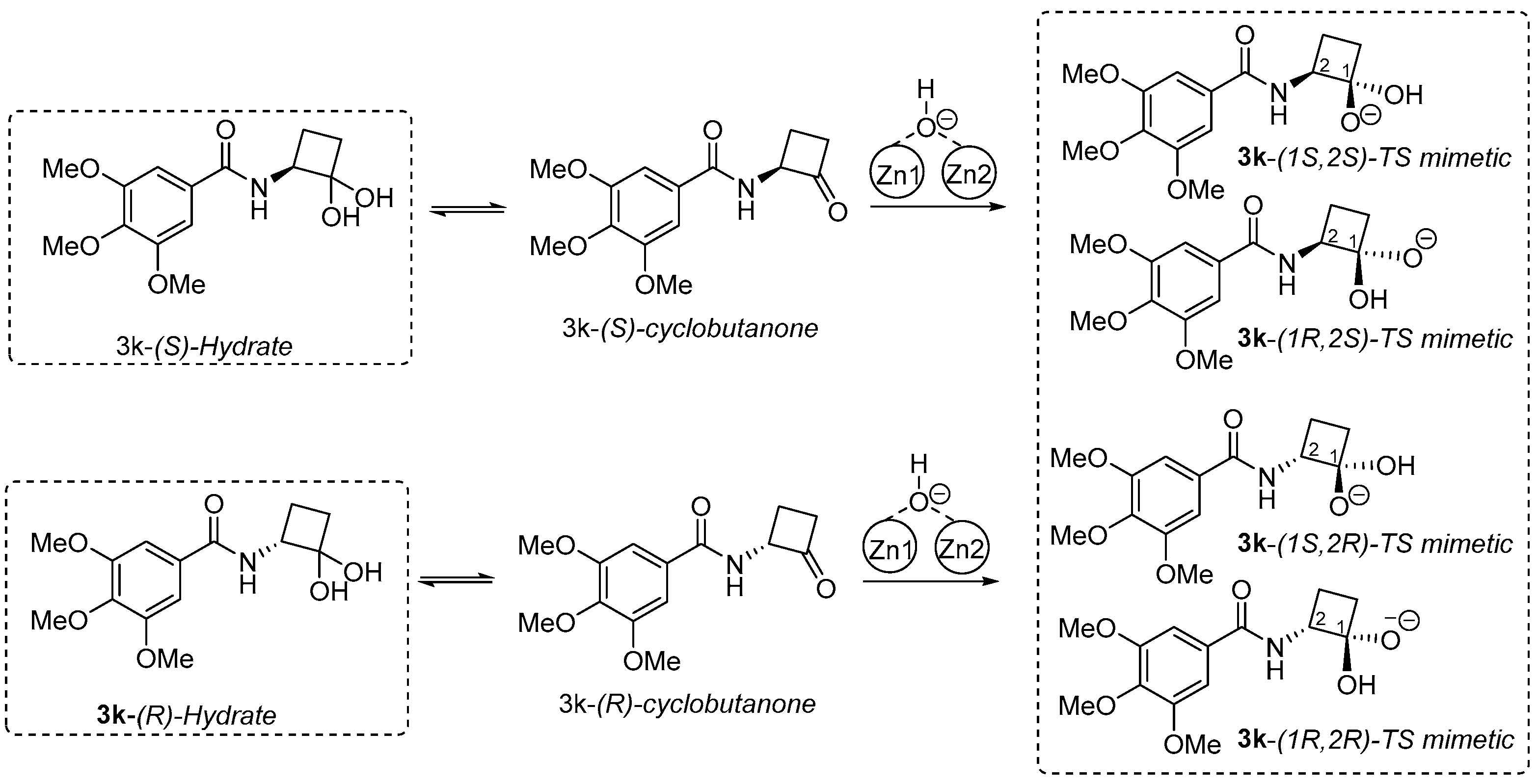
3,4,5-Trimethoxy-N-(2-oxocyclobutyl)benzamide (3k). Acetal (±)-2 (30 mg, 0.18 mmol) was suspended in methylene chloride (0.9 mL) and cooled to 0 °C. Triethylamine (49.9 µL, 0.36 mmol) was then added to the solution, followed by dropwise addition of 3,4,5-trimethoxybenzoyl chloride (41.5 mg, 0.18 mmol). The reaction was allowed to stir for 5 h at room temperature with periodic TLC (hexane/diethyl ether = 50/50) and HPLC monitoring. Upon completion, the reaction mixture was successively washed with 1 M HCl (3 mL) and water (3 mL). Then, this methylene chloride layer containing the acetal intermediate was subjected to hydrolysis by vigorously stirring with 1 M HCl (1 mL) overnight. After the hydrolysis was determined to be complete via TLC and HPLC, the organic layer was successively washed with water (3 mL) and brine (3 mL) and dried over Na2SO4. Then the solvent was removed under reduced pressure, and the resultant crude product was purified via column chromatography using ethyl acetate/hexane (50/50) to afford compound 3k as a white solid (25 mg, 21%): mp 144–146 °C. 1H NMR (500 MHz, CDCl3) δ 6.99 (s, 2H), 6.90 (NH, d, J = 7.6 Hz, 1H), 5.11–5.02 (m, 1H), 3.87 (d, J = 1.9 Hz, 9H), 3.10–3.01 (m, 1H), 3.04–2.94 (m, 1H), 2.51 (dtd, J = 11.0, 9.7, 5.6 Hz, 1H), 2.22 (dddd, J = 11.1, 10.0, 9.1, 8.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 206.1, 166.6, 153.1, 141.3, 128.3, 104.5, 77.3, 77.1, 76.8, 64.7, 60.9, 56.3, 42.2, 19.6. HRMS (ESI) calcd for M+Na+ C14H17NO5: 302.0999, found (M+Na+) 302.0990.
4.2.2. General Procedure for N-Heterocyclic Benzamide Cyclobutanones (3l-3r)
The general procedure for the synthesis of benzamide cyclobutanones (3l-3r) was followed. In order to neutralize and isolate the desired product as the free base, the hydrolysis reaction was quenched by adding saturated Na2CO3 until effervescence ceased. The organic product was extracted using ethyl acetate (3 × 3 mL) or methylene chloride for highly water-soluble analogs, and the combined organic layers were dried over Na2SO4. Then, the solvent was evaporated under reduced pressure, and the resultant crude product was purified via column chromatography to afford the heterocyclic benzamide cyclobutanones.
N-(2-Oxocyclobutyl)picolinamide (3l). The crude product of 3l was purified via column chromatography using ethyl acetate/hexane (40/60) to afford compound 3l as a white solid (37.7 mg, 79.5%): mp 97–99 °C. 1H NMR (500 MHz, CDCl3) δ 8.56 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 8.50 (NH, d, J = 8.1 Hz, 1H), 8.18 (dt, J = 7.8, 1.1 Hz, 1H), 7.87 (td, J = 7.7, 1.7 Hz, 1H), 7.46 (ddd, J = 7.6, 4.8, 1.3 Hz, 1H), 5.22 (dt, J = 9.9, 7.8 Hz, 1H), 3.05 (dd, J = 9.7, 7.9 Hz, 2H), 2.56 (tt, J = 10.6, 7.1 Hz, 1H), 2.24 (dtd, J = 11.0, 9.6, 8.3 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.2, 164.1, 149.0, 148.2, 137.4, 126.6, 122.4, 64.1, 42.3, 19.6. HRMS (ESI) calcd for MNa+ C10H10N2O2: 213.0634, found (M+Na+) 213.0630.
N-(2-Oxocyclobutyl)nicotinamide (3m). The crude product of 3m was purified via column chromatography using methanol/ethyl acetate (1/99) to afford compound 3m as a white solid (25.3 mg, 53%): 1H NMR (500 MHz, CDCl3)δ 8.98 (dd, J = 2.3, 0.8 Hz, 1H), 8.68 (dd, J = 4.9, 1.7 Hz, 1H), 8.12 (dt, J = 8.0, 2.0 Hz, 1H), 7.68 (d, J = 7.7 Hz, 1H), 7.37 (ddd, J = 8.0, 4.9, 0.9 Hz, 1H), 5.13 (dt, J = 10.3, 7.9 Hz, 1H), 3.01 (dd, J = 9.4, 7.7 Hz, 2H), 2.51 (tt, J = 10.7, 7.1 Hz, 1H), 2.24 (dtd, J = 11.0, 9.6, 8.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.8, 165.2, 152.3, 148.0, 135.6, 129.1, 123.7, 64.4, 42.2, 19.4. HRMS (ESI) calcd for MH+ C10H10N2O2: 191.0815, found (M+Na+) 191.0810.
2-Chloro-N-(2-oxocyclobutyl)nicotinamide (3n). The crude product of 3n was purified via column chromatography using ethyl acetate/hexane (70/30) to afford compound 3n as a white crystalline solid (25.3 mg, 64%): mp 80–82 °C. 1H NMR (500 MHz, CDCl3) δ 8.48 (dd, J = 4.7, 2.0 Hz, 1H), 8.14 (dd, J = 7.7, 2.0 Hz, 1H), 7.37 (dd, J = 7.6, 4.7 Hz, 1H), 7.18 (NH, s, 1H), 5.20–5.11 (m, 1H), 3.11–3.03 (m, 2H), 2.64–2.53 (m, 1H), 2.23 (dtd, J = 11.1, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 204.0, 164.1, 151.4, 147.2, 140.2, 129.9, 122.9, 77.3, 77.01, 76.6, 64.5, 42.5, 19.5. HRMS (ESI) calcd for M+Na+ C10H9ClN2O2: 247.0245, found 247.024 (35Cl), 249.021 (37Cl).
5-Bromo-N-(2-oxocyclobutyl)nicotinamide (3o). The crude product of 3o was purified via column chromatography using ethyl acetate/hexane (50/50) to afford compound 3o as a white crystalline solid (25.3 mg, 64%). %): mp 114–115 °C. 1H NMR (500 MHz, CDCl3) δ 8.89 (d, J = 2.3 Hz, 1H), 8.80 (d, J = 2.2 Hz, 1H), 8.27 (t, J = 2.1 Hz, 1H), 7.06 (NH, d, J = 7.4 Hz, 1H), 5.18 (dddd, J = 11.0, 9.5, 8.0, 1.6 Hz, 1H), 3.14–2.99 (m, 2H), 2.58 (tdd, J = 10.8, 8.4, 5.8 Hz, 1H), 2.27–2.15 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 204.7, 163.7, 153.7, 145.9, 138.0, 130.3, 121.0, 77.3, 77.0, 76.8, 64.4, 42.4, 19.6. HRMS (ESI): Calcd for (MH+) C10H10BrN2O2: 268.9920, found 268.9916 (79Br), 269.9948, 270.9896 (81Br).
N-(2-Oxocyclobutyl)isonicotinamide (3p). The crude product of 3p was purified via column chromatography using methanol/ethyl acetate (1/99) to afford compound 3p as a white solid (15.4 mg, 33%): mp 119–121 °C. 1H NMR (500 MHz, CDCl3) δ 8.77–8.72 (m, 2H), 7.64–7.59 (m, 2H), 7.10 (NH, d, J = 7.4 Hz, 1H), 5.22–5.12 (m, 1H), 3.13–2.98 (m, 2H), 2.64–2.52 (m, 1H), 2.20 (dtd, J = 11.1, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 204.7, 165.0, 150.6, 140.3, 120.9, 77.3, 77.0, 76.8, 64.4, 42.4, 19.6. HRMS (ESI) calcd for MH+ C10H10N2O2: 191.0815, found 191.0810.
N-(2-Oxocyclobutyl)quinoline-3-carboxamide (3q). The crude product of 3q was purified via column chromatography using ethyl acetate/hexane (80/20) to afford compound 3q as a white solid (30.0 mg, 50%): mp 155–156 °C. 1H NMR (500 MHz, CDCl3) δ 9.18 (d, J = 2.3 Hz, 1H), 8.52 (dd, J = 2.3, 0.8 Hz, 1H), 8.04 (dd, J = 8.4, 1.1 Hz, 1H), 7.79 (dd, J = 8.2, 1.4 Hz, 1H), 7.73 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.53 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.21 (NH, s, 1H), 5.15 (dt, J = 9.9, 7.7 Hz, 1H), 2.99 (dd, J = 9.4, 7.8 Hz, 2H), 2.57–2.46 (m, 1H), 2.19 (dtd, J = 11.1, 9.6, 8.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.3, 165.2, 149.3, 148.0, 136.2, 131.6, 129.3, 128.9, 127.7, 126.8, 125.8, 77.3, 77.0, 76.8, 64.6, 42.4, 19.7. HRMS (ESI) calcd for MNa+ C14H12N2O2: 263.0791, found (M+Na+) 263.0790.
N-(2-Oxocyclobutyl)quinoline-8-carboxamide (3r). The crude product of 3r was purified via column chromatography using ethyl acetate/hexane (70:30) to afford compound 3r as a white soft solid (11.8 mg, 19%): mp 89–91 °C. 1H NMR (500 MHz, CDCl3) δ 11.83 (NH, d, J = 7.3 Hz, 1H), 8.93 (dd, J = 4.3, 1.8 Hz, 1H), 8.83 (dd, J = 7.4, 1.6 Hz, 1H), 8.29 (dd, J = 8.4, 1.8 Hz, 1H), 7.99 (dd, J = 8.1, 1.6 Hz, 1H), 7.68 (dd, J = 8.1, 7.3 Hz, 1H), 7.51 (dd, J = 8.3, 4.3 Hz, 1H), 5.25–5.15 (m, 1H), 3.16 (dddd, J = 17.5, 10.1, 4.6, 2.7 Hz, 1H), 3.04 (dddd, J = 17.6, 10.6, 8.8, 2.0 Hz, 1H), 2.56 (qd, J = 10.5, 4.7 Hz, 1H), 2.39 (dddd, J = 11.0, 10.1, 8.8, 8.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 206.3, 165.7, 149.4, 145.5, 137.8, 134.2, 132.4, 128.5, 127.8, 126.6, 121.0, 65.0, 42.3, 19.7. HRMS (ESI) calcd for MNa+ C14H12N2O2: 263.0791, found (M+Na+) 263.0810.
2-(3-Methoxyphenyl)-N-(2-oxocyclobutyl)acetamide (3s). The crude product of 3s was purified via column chromatography using ethyl acetate/hexane (40/60) where compound 3s was isolated as an off-white crystalline solid (26 mg, 88% HPLC purity). Recrystallization of the above-mentioned solid from hot ethyl acetate afforded compound 3s as white crystals (11.4 mg, 20%): mp 104–105 °C. 1H NMR (500 MHz, CDCl3) δ 7.27 (dd, J = 8.3, 7.4 Hz, 1H), 6.87–6.81 (m, 2H), 6.80 (t, J = 2.1 Hz, 1H), 5.94 (NH, d, J = 7.4 Hz, 1H), 4.86 (dtd, J = 9.6, 7.4, 6.7, 1.2 Hz, 1H), 3.81 (s, 3H), 3.56 (s, 2H), 2.99–2.84 (m, 2H), 2.44–2.33 (m, 1H), 2.02 (dtd, J = 11.0, 9.6, 8.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.2, 170.5, 160.1, 135.7, 130.2, 121.7, 115.0, 113.1, 64.2, 55.3, 43.2, 42.01, 19.5. HRMS (ESI) calcd for MNa+ C13H15NO3: 256.0944, found (M+Na+) 256.095.
2-(4-Bromophenyl)-N-(2-oxocyclobutyl)acetamide (3t). The crude product of 3t was purified via column chromatography using ethyl acetate/hexane (50/50) isocratic elution to afford compound 3t as a white crystalline solid (49.7 mg, 71%): mp 124–127 °C. 1H NMR (500 MHz, CDCl3) δ 7.51–7.43 (m, 2H), 7.20–7.11 (m, 2H), 5.95 (d, 1H), 4.89 (dt, J = 10.3, 8.0 Hz, 1H), 3.53 (s, 2H), 3.02–2.86 (m, 2H), 2.40 (tt, J = 10.5, 7.2 Hz, 1H), 2.01 (dtd, J = 11.1, 9.5, 8.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.1, 170.0, 133.2, 132.2, 131.1, 121.6, 64.2, 42.4, 42.1, 19.5. HRMS (ESI) calcd for MNa+ C12H12BrNO2: 303.9944, found (M+Na) 303.994 (100%) 304.997 (13.56%).
3-(1H-Benzo[d]imidazol-2-yl)-N-(2-oxocyclobutyl)propenamide (3u). 2-Benzimidazolepropionic acid was reacted with acetal (±)-2 following the general T3P coupling reaction method. Upon the completion of the acetal hydrolysis, acetone was evaporated off under reduced pressure. Then, the reaction was quenched by adding saturated Na2CO3 until effervescence ceased. The organic product was extracted using methylene chloride (5 × 2 mL) and the combined organic layers were dried over Na2SO4. Then, the solvent was evaporated under reduced pressure, and the resultant crude product was purified via column chromatography using methanol/ethyl acetate (3/97) isocratic elution to afford an off-white solid with 93% HPLC purity. This solid was washed with ethyl acetate (3 × 0.5 mL) to yield cyclobutanone 3u as a white crystalline solid (10.5 mg, 16%): mp 179–181 °C. 1H NMR (500 MHz, CDCl3) δ 7.54 (dt, J = 6.7, 3.3 Hz, 2H), 7.26 (NH, s, 1H), 7.21 (dt, J = 7.0, 3.5 Hz, 2H), 7.14 (NH, d, J = 7.6 Hz, 1H), 4.78 (tdd, J = 7.7, 5.9, 4.2 Hz, 1H), 3.27–3.13 (m, 2H), 3.03–2.87 (m, 2H), 2.82–2.62 (m, 2H), 2.44–2.31 (m, 1H), 2.12 (tt, J = 10.0, 9.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 205.8, 172.6, 153.7, 122.4, 64.4, 42.1, 33.4, 24.6, 19.1. HRMS (ESI) calcd for MH+ C14H16N3O2: 258.1237, found (MH+) 258.1230.
4.2.3. General Procedure for Synthesis of N-Aryl Sulfonamide-Amino Acid Intermediates
Following the general method reported by Mishra [
36], to a vigorously stirring solution of amino acid (1.21 mmol) in DI water (1.5 mL), sodium carbonate (179.8 mg, 1.45 mmol) was added. Once all the solutes were dissolved, the solution was cooled to 0 °C, and the respective aryl sulfonyl chloride (1.45 mmol) was added to the stirring reaction mixture in four portions over 1 h. Then, the reaction was allowed to warm up to rt, and the slurry was stirred at rt for 4 h or until completion. The reaction progress was monitored via TLC (methanol/methylene chloride = 10/90). Once completed, the mixture was acidified using 2 N HCl until the pH of the solution was reduced to pH 2. Crystals formed during the acidification were filtered and washed with pH 2.2 buffer. Final pure products (
3v-
3x) were dried under high vacuum. Both proton and carbon NMR spectra of pure products
3v-
3x matched the reported characterization data as indicated.
4.2.4. General Procedure for the Synthesis of Amino Acid Cyclobutanone Analogs 3v-3ae
To a solution of substituted N-benzene sulfonamide amino acids (1 eq) and acetal (±)-2 (1.2 eq) in ethyl acetate (0.2 M), N-methyl morpholine (4.0 eq) was added. Then, propylphosphonic anhydride (T3P, 2.5 eq) was added to the above mixture as a solution in ethyl acetate (purchased as ≥50 weight % in EA). The reaction mixture was stirred under N2 at rt to 40 °C for 18 h or until complete consumption of protected carboxylic acid intermediates determined with HPLC analysis. The reaction was quenched by adding water (3 mL) and then the organic product was extracted using ethyl acetate (3 × 3 mL). The combined organic layers were washed successively with water (3 × 3 mL) and 1N HCl (3 mL) and then dried over Na2SO4. The solvent was removed via evaporation under reduced pressure, providing the corresponding acetal intermediate. Without further purification, crude acetal (1 eq, final concentration was 0.1 M) was subjected to hydrolysis conditions, wherein the crude mixture was dissolved in acetone, water (10% v/v), and 1N HCl (30% v/v). Then, the reaction mixture was stirred at 40 °C overnight with periodic HPLC monitoring. Upon completion, the organic product was extracted using ethyl acetate (3 × 3 mL) or methylene chloride for highly water-soluble analogs, and the combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure, and the crude mixture was purified via column chromatography to afford the corresponding amino acid-derived cyclobutanone (3v-3ae).
(2S)-2-((4-Methylphenyl)sulfonamido)-N-(2-oxocyclobutyl)-3-phenylpropanamide (
3v). Tosyl-L-phenylalanine (50.0 mg, 0.129 mmol) was prepared according to Mishra [
36] and was employed in the reaction (50.0 mg, 0.129 mmol). The crude mixture of
3v was purified via column chromatography eluting with a gradient of ethyl acetate/hexane (20/80) and ethyl acetate/hexane (40/60) followed by recrystallization from hot EA to afford compound
3v (9.8 mg, 20%) as white crystalline solid.
1H NMR (500 MHz, CDCl
3) δ 7.57–7.44 (m, 2H), 7.25–7.15 (m, 5H), 6.96–6.87 (m, 2H), 6.72 (d, J = 8.3 Hz, 1H), 4.97 (dtt, J = 10.1, 8.2, 2.1 Hz, 1H), 4.85 (d, J = 6.6 Hz, 1H), 3.85 (dt, J = 7.3, 6.4 Hz, 1H), 3.01–2.89 (m, 3H), 2.88 (dddd, J = 17.4, 9.9, 4.7, 2.3 Hz, 1H), 2.43 (s, 3H), 2.46–2.35 (m, 1H), 1.86 (dtd, J = 11.1, 9.7, 8.3 Hz, 1H). HRMS (ESI): Calcd for (MH
+) C
20H
23N
2O
4S: 387.1373, found 387.1366.
(2R)-2-((4-Methylphenyl)sulfonamido)-N-(2-oxocyclobutyl)-3-phenylpropanamide (
3w). Tosyl-D-phenylalanine (50.0 mg, 0.129 mmol) was prepared according to Mishra [
36] and was employed in the reaction. The crude mixture of
3w was purified via column chromatography eluting with a gradient of ethyl acetate/hexane (20/80) and ethyl acetate/hexane (30/70) to afford compound
3w (18.1 mg, 36.2%) as a white solid: mp 190–192 °C.
1H NMR (500 MHz, CDCl
3) δ 7.57–7.48 (m, 2H), 7.24–7.14 (m, 5H), 6.95–6.86 (m, 2H), 6.75 (NH, d, J = 8.3 Hz, 1H), 4.98 (dtt, J = 10.2, 8.3, 1.9 Hz, 0.5H), 4.87 (NH, d, J = 6.6 Hz, 0.5H), 4.82 (NH, d, J = 6.8 Hz, 0.5H), 4.78–4.68 (m, 0.5H), 3.87 (dtd, J = 16.8, 7.0, 6.1 Hz, 1H), 3.07–2.93 (m, 1.5H), 2.96–2.91 (m, 1H), 2.93–2.85 (m, 1H), 2.83 (dd, J = 14.1, 5.8 Hz, 0.5H), 2.44 (s, 1.5H), 2.43 (s, 1.5H), 2.43–2.36 (m, 0.5H), 2.39–2.31 (m, 0.5H), 2.16 (dddd, J = 11.2, 10.3, 8.9, 8.0 Hz, 0.5H), 1.86 (dtd, J = 11.2, 9.7, 8.4 Hz, 0.5H).
13C NMR (126 MHz, CDCl
3) δ 204.5, 204.4, 170.2, 169.9, 144.2, 144.1, 135.4, 135.3, 134.9, 134.7, 130.0, 129.9, 129.3, 129.1, 129.0, 127.4, 127.4, 127.2, 127.2, 77.3, 77.0, 76.8, 64.3, 63.7, 57.6, 57.3, 42.3, 42.1, 38.2, 37.7, 21.6, 21.6, 19.4, 18.9.
(2S)-3-(1H-Indol-3-yl)-2-((4-methylphenyl)sulfonamido)-N-(2-oxocyclobutyl)propanamide (
3x). Tosyl-L-tryptophan was prepared according to Mishra [
36] and was employed in the reaction (50.0 mg, 0.139 mmol). The crude mixture of
3x was purified via column chromatography on a Teledyne Isco Rf Flash chromatography unit eluting with a gradient of ethyl acetate/hexane (20/80), ethyl acetate/hexane (30/70), and ethyl acetate/hexane (40/60) to afford compound
3x (12.4 mg, 21%) as an off-white solid: mp 200–201 °C.
1H NMR (500 MHz, acetonitrile-
d3) δ 9.07 (NH, s, 1H), 7.48–7.41 (m, 2H), 7.41–7.32 (m, 2H), 7.16–7.09 (m, 3H), 7.10 (NH, d,
J = 7.5 Hz, 1H), 7.07–6.96 (m, 2H), 5.82 (NH, s, 1H), 4.68 (dtt,
J = 10.2, 8.0, 2.2 Hz, 1H), 3.90 (dd,
J = 8.1, 5.7 Hz, 1H), 3.14–3.06 (m, 1H), 2.95–2.73 (m, 3H), 2.35 (s, 3H), 2.21–2.10 (m, 1H), 1.82 (tt,
J = 10.5, 8.6 Hz, 1H).
13C NMR (126 MHz, CD
3CN) δ 205.6, 170.7, 143.5, 136.6, 136.4, 129.4, 127.1, 126.6, 126.5, 124.1, 121.4, 118.9, 111.3, 109.0, 63.6, 63.6, 57.0, 41.4, 41.3, 28.7, 28.6, 20.6, 20.6, 18.2, 18.1. HRMS (ESI): Calcd for (MH
+) C
22H
24N
3O
4S: 426.1482, found 426.1472.
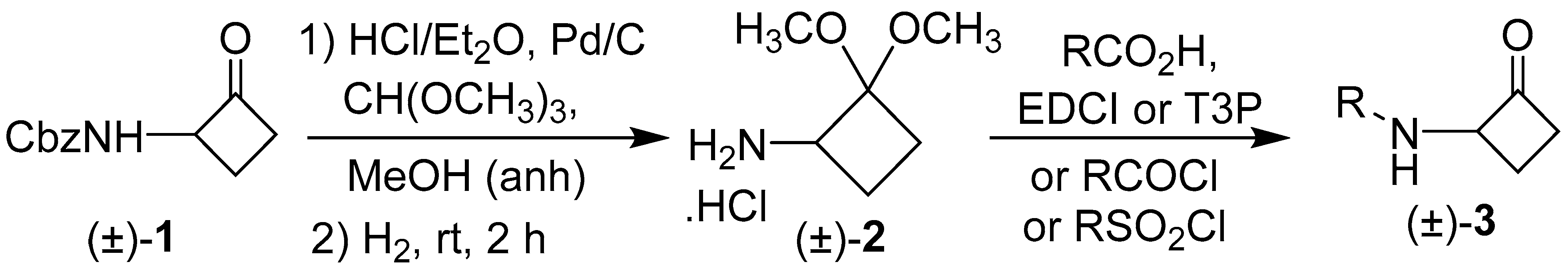
General procedure of D-valine sulfonamide para-phenyl substituents (3y, 3aa-3ac) Valine tert-butyl ester HCl salt (0.477 mmol) was dissolved in anhydrous pyridine (1.0 mL) and stirred until all the solutes were dissolved. The reaction mixture was cooled to 0 °C with an ice bath, and the respective para-benzene sulfonyl chloride (0.477 mmol) was added to the stirring reaction mixture. Then, the reaction was allowed to warm up to rt and stirred at rt for 24 h with periodic monitoring using HLPC. Upon completion, the reaction mixture was diluted with methylene chloride (3 mL), and the organic layer was washed successively with water (3 mL), 1N HCl (10 × 3 mL), and brine (3 mL) and then dried over Na2SO4. The solvent was removed via evaporation under reduced pressure providing a pale-yellow solid. Without further purification, the respective phenyl-D-valine tert butyl esters were subjected to hydrolysis conditions. To a solution of crude phenyl-D-valine tert-butyl ester (0.188 mmol) in methylene chloride (0.94 mL), TFA (282 µL, 30% v/v) was added under nitrogen. The mixture was stirred at room temperature overnight and monitored via TLC (EA/hexane = 50/50). The solvent was evaporated under reduced pressure. To the resultant residue, toluene (2 mL) was added to enable the formation of an azeotrope. The excess TFA was removed via evaporation on a rotary evaporator at 50 °C providing a yellow crystalline solid with yields between 90–99%. The intermediates were immediately subjected to coupling conditions to form cyclobutanones 3y, 3aa-3ac.
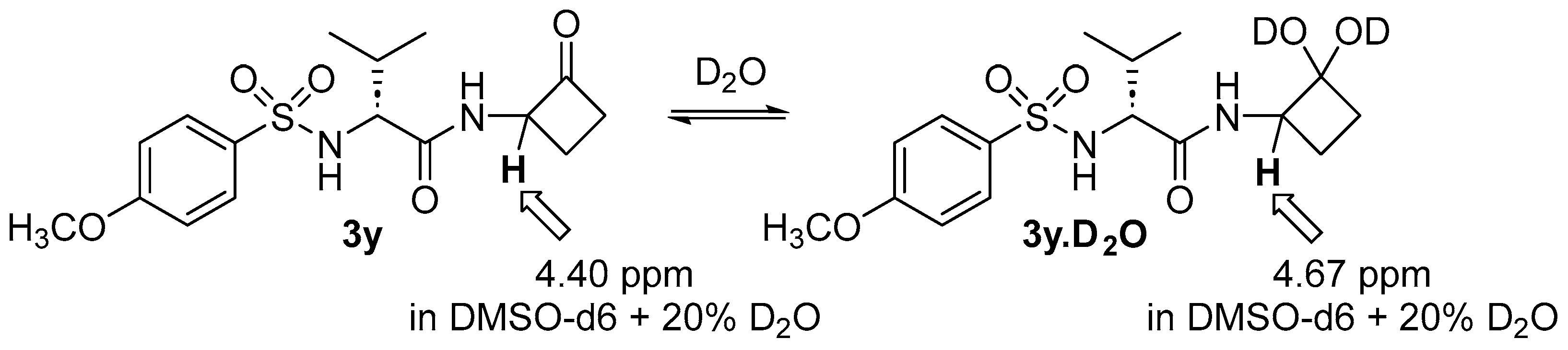
(2R)-2-((4-Methoxyphenyl)sulfonamido)-3-methyl-N-(2-oxocyclobutyl)butanamide (
3y). D-Cyclobutanone
3y was synthesized from the intermediate [
37] described in the general procedure of D-valine sulfonamide para-phenyl substituents. Compound
3y was synthesized from this intermediate (30 mg and 0.104 mmol) following the general procedure of the T3P coupling reactions of N-Ts amino acids with 2-aminocyclobutanone synthon
(±)-2, and the acetal intermediate was subjected to hydrolysis using general reaction conditions (1 M HCl, acetone, and H
2O) and stirred at 40 °C for 18 h. The crude mixture of
3y was purified via column chromatography using ethyl acetate/hexane (60/40) to afford Ts-D-valine cyclobutanone analog
3y (15.5 mg, 42%) as a white solid: mp 179–181.
1H NMR (500 MHz, CDCl
3) δ 7.84–7.77 (m, 2H), 7.04–6.96 (m, 2H), 6.51 (dd, J = 16.8, 7.7 Hz, 1H), 5.08 (dd, J = 13.7, 7.8 Hz, 1H), 4.89–4.76 (m, 1H), 3.90 (s,1.5H), 3.89 (s, 1.5H), 3.48 (ddd, J = 16.5, 7.8, 5.1 Hz, 1H), 2.99–2.87 (m, 2H), 2.43–2.30 (m, 1H), 2.17–2.06 (m, 1H), 2.01–1.94 (m, 0.5H), 1.87 (dtd, J = 11.0, 9.6, 8.1 Hz, 0.5H), 0.87 (dd, J = 6.9, 3.2 Hz, 3H), 0.81 (t, J = 6.7 Hz, 3H).
13C NMR (126 MHz, CDCl
3) δ 204.2, 170.4, 170.3, 163.3, 130.5, 129.6, 114.4, 64.1, 63.9, 61.8, 61.8, 55.7, 42.3, 42.2, 31.1, 31.1, 19.2, 19.0, 17.2, 17.1. HRMS (ESI): Calcd for (MH+) C
16H
23N
2O
5S: 355.1322, found 355.1309.
(2S)-2-((4-Methoxyphenyl)sulfonamido)-3-methyl-N-(2-oxocyclobutyl)butanamide (
3z). The starting material ((4-methoxyphenyl)sulfonyl)-
L-valine was synthesized as described by Marques et al. [
37], and the
1H and
13C NMR matched spectra reported in the literature [
38]. Without further purification, ((4-methoxyphenyl)sulfonyl)-
L-valine was coupled with the 2-aminocyclobutanone synthon following the general procedure of the T3P coupling reactions of
N-Ts amino acids. The acetal intermediate was subjected to hydrolysis using general reaction conditions (1 M HCl, acetone, and H
2O) and stirred at 40 °C for 18 h to afford (2
S)-2-((4-methoxyphenyl)sulfonamido)-3-methyl-
N-(2-oxocyclobutyl)butanamide. Further purification on the crude mixture was conducted via rinsing the solid with ether three times to afford analog
3z (15.3 mg, 15%) as a white solid: mp 179–181 °C.
1H NMR (500 MHz, CHCl
3) δ 7.75–7.67 (m, 2H), 6.94–6.85 (m, 2H), 6.42 (dd, J = 15.7, 7.6 Hz, 1H), 4.99 (dd, J = 11.8, 7.8 Hz, 1H), 4.79–4.66 (m, 1H), 3.81 (d, J = 5.0 Hz, 3H), 3.39 (ddd, J = 16.3, 7.8, 5.1 Hz, 1H), 2.91–2.78 (m, 2H), 2.34–2.21 (m, 1H), 2.08–1.96 (m, 1H), 1.94–1.72 (m, 1H), 0.78 (dd, J = 6.9, 3.5 Hz, 3H), 0.72 (t, J = 6.7 Hz, 3H).
13C NMR (126 MHz, CDCl
3) δ 204.2, 170.4, 170.3, 163.3, 130.6, 129.6, 114.4, 64.1, 63.9, 61.8, 55.7, 42.3, 42.2, 31.1, 31.1, 19.2, 19.1, 17.1, 17.1.
(2R)-2-((4-Cyanophenyl)sulfonamido)-3-methyl-N-(2-oxocyclobutyl)butanamide (3aa). Cyclobutanone 3aa was synthesized from the intermediate described in the general procedure of D-valine sulfonamide para-phenyl substituents following the general procedure of the T3P coupling reactions of N-Ts amino acids with 2-aminocyclobutanone synthon (±)-2 followed by immediate acid-hydrolysis of the acetal to form the ketone. The crude mixture of 3aa was purified via column chromatography using ethyl acetate/hexane (60/40) to afford Ts-D-valine cyclobutanone analog 3aa (23.1 mg, 30%) as a white solid: mp 149.5–151 °C. 1H NMR (500 MHz, chloroform-d) 7.92–7.84. (m, 2H), 7.79–7.70 (m, 2H), 5.29 (dd, J = 24.2, 8.8 Hz, 1H), 3.47 (ddd, J = 11.3, 8.9, 5.3 Hz, 1H), 2.95–2.85 (m, 1H), 2.85–2.77 (m, 1H), 2.35–2.22 (m, 1H), 1.99–1.89 (m, 1H), 1.70 (dtd, J = 11.1, 9.7, 8.2 Hz, 1H), 0.87–0.79 (m, 6H). 13C NMR (126 MHz, chloroform-d) δ 203.5, 169.5, 132.9, 127.9, 117.2, 63.8, 61.9, 42.3, 31.8, 19.4, 19.1, 17.3.
Methyl 4-(N-((2R)-3-methyl-1-oxo-1-((2-oxocyclobutyl)amino)butan-2-yl)sulfamoyl)benzoate (3ab). Cyclobutanone sulfonamide 3ab was synthesized from the intermediate described in the general procedure of D-valine sulfonamide para-phenyl substituents following the general procedure of the T3P coupling reactions of N-Ts amino acids with 2-aminocyclobutanone synthon (±)-2 followed by immediate acid-hydrolysis of the acetal to form the ketone. The crude cyclobutanone 3ab was purified via column chromatography using ethyl acetate/hexane (60/40) to afford Ts-D-valine cyclobutanone analog 3ab (23.8 mg, 20%) as a white solid: mp 190–191 °C. 1H NMR (500 MHz, chloroform-d) δ 8.2 (dq, J = 8.5, 1.9 Hz, 2H), 7.9 (dq, J = 8.5, 2.0 Hz, 2H), 5.3 (s, 1H), 4.9–4.7 (m, 1H), 3.9 (d, J = 2.1 Hz, 3H), 3.6–3.5 (m, 1H), 3.0–2.9 (m, 2H), 2.4–2.3 (m, 1H), 2.1 (hd, J = 6.8, 5.3 Hz, 1H), 1.8 (ddtd, J = 35.8, 11.1, 9.6, 8.1 Hz, 1H), 0.9 (ddd, J = 14.4, 6.8, 3.1 Hz, 6H). 13C NMR (126 MHz, chloroform-d) δ 203.8, 169.8, 165.6, 143.3, 134.2, 130.4, 130.4, 127.4, 127.3, 63.9, 61.9, 52.7, 42.3, 31.5, 19.3, 19.0, 17.2.
(2R)-2-((4-hydroxyphenyl)sulfonamido)-3-methyl-N-(2-oxocyclobutyl)butanamide (3ac). Cyclobutanone sulfonamide 3ac was synthesized from the intermediate described in the general procedure of D-valine sulfonamide para-phenyl substituents following the general procedure of the T3P coupling reactions of N-Ts amino acids with 2-aminocyclobutanone synthon (±)-2 followed by immediate acid-hydrolysis of the acetal to form the ketone. The crude cyclobutanone 3ac was purified via column chromatography using ethyl acetate/hexane (60/40) to afford Ts-D-valine cyclobutanone analog 3ac (11 mg, 14%) as a white solid: mp 139–141 °C. 1H NMR (500 MHz, chloroform-d) δ 7.66–7.62 (m, 2H), 7.2 (d, J = 2.1 Hz, 2H), 6.9–6.8 (m, 1H), 5.0 (dtt, J = 10.3, 8.3, 2.1 Hz, 2H), 3.7–3.6 (m, 1H), 3.0–2.9 (m, 1H), 2.9 (t, J = 2.0 Hz, 1H), 2.5–2.5 (m, 1H), 2.1–2.0 (m, 1H), 1.2 (s, 6H). 13C (126 MHz, chloroform-d) 208.27, 173.76, 159.37, 131.28, 120.62, 63.40, 42.76, 31.94, 22.70, 19.28, 14.13.
4.2.5. Synthesis of Cbz Protected Amino Acid Cyclobutanone Analogs 3ad-3ag
Cbz protected amino acids (Phe, Try, Val, 50 mg) were coupled with 2-aminocyclobutanone synthon (±)-2 following the general T3P coupling reaction procedure. The resulting acetal intermediates were subjected to hydrolysis without further purification. To a solution of crude acetal product (1 eq, concentration = 0.1 M) in THF (40% v/v), water (20% v/v) and acetic acid were added, and the reaction mixture was stirred at 60 °C overnight or until deemed complete via HPLC. Upon completion, excess acetic acid was neutralized by adding a solution of saturated Na2CO3 until a pH of 8 was achieved, and the organic product was extracted with methylene chloride (3 × 3 mL). The combined organic layers were dried over Na2SO4, and the solvent was evaporated under reduced pressure. The crude mixture was purified via column chromatography to afford the corresponding Cbz-protected amino acid-derived cyclobutanone (3ad-3af).
Benzyl ((2S)-1-oxo-1-((2-oxocyclobutyl)amino)-3-phenylpropan-2-yl)carbamate (3ad). The crude mixture of 3ad was purified via column chromatography eluting with a gradient of ethyl acetate/hexane (20/80) to (30/70). Appropriate fractions were combined and concentrated to produce 3ad as a white solid (94% HPLC purity) which was then recrystallized from hot methylene chloride to produce white needle-like crystals of compound 3ad with 97% HPLC purity (30.2 mg, 49%): mp 100–101 °C. 1H NMR (500 MHz, CDCl3) δ 7.40–7.23 (m, 8H), 7.23–7.17 (m, 2H), 6.63 (NH, d, J = 7.6 Hz, 0.5H), 6.48 (NH, d, J = 7.9 Hz, 0.5H), 5.46 (t, J = 10.6 Hz, 1H), 5.07 (t, J = 3.3 Hz, 2H), 4.96–4.87 (m, 0.5H), 4.78 (qt, J = 7.9, 1.4 Hz, 0.5H), 4.45 (d, J = 7.0 Hz, 1H), 3.09 (d, J = 6.7 Hz, 2H), 2.98–2.83 (m, 2H), 2.38 (dd, J = 10.9, 5.2 Hz, 0.5H), 2.33 (dd, J = 10.3, 4.7 Hz, 0.5H), 2.03 (q, J = 9.6 Hz, 0.5H), 1.90–1.73 (m, 0.5H). 13C NMR (126 MHz, CDCl3) δ 204.8, 204.8, 170.8, 170.7, 156.0, 136.2, 136.2, 136.1, 129.4, 129.4, 128.8, 128.6, 128.3, 128.0, 127.1, 67.2, 64.0, 63.7, 56.0, 55.8, 42.2, 42.1, 38.6, 38.4, 19.4, 19.1. HRMS (ESI): Calcd for (MH+) C21H23N2O4: 367.1652, found 367.1646.
Benzyl ((2R)-1-oxo-1-((2-oxocyclobutyl)amino)-3-phenylpropan-2-yl)carbamate (3ae). The crude mixture of 3ae was purified via column chromatography eluting with a gradient of ethyl acetate/hexane (20/80) and ethyl acetate/hexane (30/70). Appropriate fractions were combined and concentrated to produce a white solid (93% HPLC purity), which was then subjected to recrystallization. The product was dissolved in a minimal amount of methylene chloride, and hexane was added dropwise until cloudiness was observed. Colorless crystals were formed providing compound 3ae (16.1 mg, 26%): mp 98–100 °C. 1H NMR (500 MHz, CDCl3) δ 7.41–7.24 (m, 8H), 7.21 (t, J = 6.0 Hz, 2H), 6.35 (d, J = 7.6 Hz, 0.5H), 6.20 (d, J = 8.0 Hz, 0.5H), 5.32 (s, 0.5H), 5.27 (s, 0.5H), 5.10 (s, 2H), 4.92 (q, J = 9.0 Hz, 0.5H), 4.81 (q, J = 8.7 Hz, 0.5H), 4.42 (q, J = 7.3 Hz, 1H), 3.17–3.11 (m, 1H), 3.06 (td, J = 13.5, 12.9, 7.6 Hz, 1H), 2.99–2.85 (m, 2H), 2.39 (dq, J = 15.8, 9.7 Hz, 1H), 2.06–2.03 (m, 0.5H), 1.88–1.81 (m, 0.5H). 13C NMR (126 MHz, CDCl3) δ 204.3, 170.7, 170.5, 156.0, 136.0, 136.0, 129.4, 129.4, 128.8, 128.6, 128.3, 128.3, 128.1, 127.2, 67.2, 64.1, 63.8, 56.0, 42.2, 42.1, 38.6, 38.2, 19.4, 19.2. HRMS (ESI): Calcd for (MH+) C21H23N2O4: 367.1652, found 367.1650.
Benzyl ((2S)-3-(4-hydroxyphenyl)-1-oxo-1-((2-oxocyclobutyl)amino)propan-2-yl)carbamate (3af). The crude mixture of 3af was purified via column chromatography eluting with a gradient of ethyl acetate/hexane (20/80) to (30/70). Combined fractions were recrystallized from hot methylene chloride to afford compound 3af (14.6 mg, 24%) as small needle-like crystals: mp 154–155 °C. 1H NMR (500 MHz, acetonitrile-d3) δ 7.42–7.26 (m, 4H), 7.23–7.13 (m, 1H), 7.11–7.04 (m, 2H), 6.77–6.71 (m, 2H), 5.88 (dd, J = 12.9, 8.5 Hz, 1H), 5.08 (d, J = 12.8 Hz, 1H), 4.99 (d, J = 12.7 Hz, 1H), 4.80 (dddd, J = 20.0, 10.0, 5.9, 2.1 Hz, 1H), 4.25 (dtd, J = 17.5, 8.7, 5.3 Hz, 1H), 3.04 (ddd, J = 13.6, 7.8, 5.4 Hz, 1H), 2.96–2.81 (m, 1H), 2.79 (dddd, J = 14.1, 12.0, 8.3, 2.7 Hz, 1H), 2.15–1.91 (m, 1H). 13C NMR (126 MHz, CD3CN) δ 206.1, 171.2, 171.1, 155.7, 137.1, 130.4, 128.5, 128.3, 128.2, 127.9, 127.5, 115.1, 66.1, 63.7, 63.6, 56.3, 56.1, 41.4, 41.4, 18.3, 18.1. HRMS (ESI): Calcd for (MNa+) C22H21N2O5Na: 405.1421, found 405.1420.
Benzyl ((2S)-3-methyl-1-oxo-1-((2-oxocyclobutyl)amino)butan-2-yl)carbamate (3ag). The crude mixture of 3ag was purified via column chromatography eluting with a gradient of ethyl acetate/hexane (20/80) to (30/70). Appropriate fractions were combined and concentrated to produce a white solid, which was then subjected to recrystallization. The product was dissolved in a minimal amount of methylene chloride, and hexane was added dropwise until cloudiness was observed. Over 24 h, needle-like crystals formed providing compound 3ag (21.3 mg, 34%): mp 140–144 °C. 1H NMR (500 MHz, CDCl3) δ 7.38 (s, 3H), 7.43–7.32 (m, 2H), 6.44–6.39 (d, 1H), 5.26 (d, J = 8.9 Hz, 1H), 5.14 (s, 2H), 4.91 (dt, J = 10.1, 7.9 Hz, 1H), 4.01 (dd, J = 8.8, 6.0 Hz, 1H), 2.98 (t, J = 8.8 Hz, 2H), 2.46 (dd, J = 13.3, 5.3 Hz, 1H), 2.18 (h, J = 6.8 Hz, 1H), 2.13–2.06 (m, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 204.4, 171.0, 152.2, 136.1, 128.6, 128.3, 128.1, 67.3, 64.2, 60.0, 42.3, 30.8, 29.7, 19.5, 19.2. HRMS (ESI): Calcd for (MH+) C17H23N2O4: 319.1652, found 319.1641.
4.2.6. General Procedure for Sulfonamide Cyclobutanones (3ah and 3ai)
Acetal (±)-2 (50.0 mg, 0.298 mmol) was suspended in methylene chloride (0.8 mL) and the reaction was cooled to 0 °C. Triethylamine (82.6 µL, 0.596 mmol) was then added to the solution, followed by dropwise addition of the requisite benzenesulfonyl chloride (87.6 mg, 0.358 mmol) in methylene chloride (0.7 mL). The reaction was stirred at 0 °C for 5 min, then allowed to warm to rt and stirred overnight. Once the reaction was deemed complete via TLC, the organic layer was washed successively with water (2 × 3 mL), 1N HCl (3 mL), and saturated Na2CO3. Then, the organic layer was dried over sodium sulfate, and the solvent was removed under reduced pressure to afford the crude acetal intermediate. Without further purification, the crude material (97.9 mg) was subjected to hydrolysis by dissolving in a mixture of acetone (2.0 mL), water (0.30 mL), and 1N HCl (0.60 mL) and was then allowed to stir at 40 °C overnight. The reaction was then partitioned between water and methylene chloride, and the organic product was extracted with methylene chloride (2 × 3 mL). The combined organic fractions were dried over Na2SO4 and concentrated under reduced pressure. The resultant crude product was purified via column chromatography to produce the corresponding sulfonamides.
N-(2-Oxocyclobutyl)-4-(trifluoromethyl)benzenesulfonamide (3ah). The crude product of 3ah was purified via column chromatography eluting with ethyl acetate/hexane (30/70) to afford benzenesulfonamide 3ah as a clear oil (60.8 mg, 70%), which formed needle-like crystals in ethyl acetate/hexane (10/90). mp 74–76 °C. 1H NMR (500 MHz, CDCl3) δ 7.97 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 5.30 (d, J = 8.1 Hz, 1H), 4.73–4.64 (m, 1H), 2.89 (dddd, J = 17.4, 11.3, 9.9, 1.7 Hz, 1H), 2.73 (dddd, J = 17.4, 9.9, 4.2, 2.5 Hz, 1H), 2.41 (tdd, J = 11.1, 10.1, 4.2 Hz, 1H), 1.79 (dtd, J = 11.3, 9.9, 8.4 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 201.30, 142.80, 133.71 (q, J C-F = 33.2 Hz), 126.50, 125.36 (q, J C-F = 3.8 Hz), (ortho carbons of aryl-CF3 long range F splitting: 123.2, 121.1, 118.9, 76.2, 76.0, 75.7, 64.8, 40.9, 19.9. HRMS (ESI): Calcd for (MH+) C11H11F3NO3S: 294.04, found 294.0405, Calcd for: (M+Na+) C11H11F3NO3SNa+: 316.0231, found 316.0222.
N-(2-Oxocyclobutyl)-4-(fluoro)benzenesulfonamide (3ai). The crude product of 3ai was purified via column chromatography on a Teledyne Isco Rf Flash chromatography unit eluting with a gradient of 100% hexane, 5% EA in hexane, and 10% EA in hexane to afford compound 3ai as a clear oil (13.6 mg, 23%). 1H NMR (500 MHz, CDCl3) δ 8.00–7.90 (m, 1H), 7.93–7.85 (m, 1H), 7.27–7.15 (m, 2H), 5.52 (d, J = 8.2 Hz, 1H), 4.72 (dtt, J = 10.3, 8.3, 2.2 Hz, 1H), 2.93 (dddt, J = 17.4, 11.3, 9.8, 1.7 Hz, 1H), 2.77 (dddd, J = 17.3, 9.9, 4.2, 2.5 Hz, 1H), 2.47–2.36 (m, 1H), 1.83 (dtd, J = 11.1, 9.9, 8.5 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 203.0, 165.23 (d, J C-F = 255.3 Hz), 136.3 (d, J C-F = 3.6 Hz), 129.8 (d, J C-F = 9.4 Hz), 116.5 (d, J C-F = 22.9 Hz), 65.9, 41.9, 20.8. HRMS (ESI) calcd for MNa+ C10H10FNO3SNa+: 266.0258, found (M+Na+) 266.0250.
(S)-N-((1-hydroxycyclopropyl)methyl)-2-((4-methylphenyl)sulfonamido)-3-phenylpropanamide (4a). Tosyl-L-phenylalanine (50 mg, 0.129 mmol) was reacted with 1-(aminomethyl)cyclopropanol (14.4 µL, 0.155 mmol) following the general T3P coupling procedure and stirred at 50 °C for 4 days. The crude mixture of 4a was purified via column chromatography on a Teledyne Isco Rf Flash chromatography unit eluting with a gradient of ethyl acetate/hexane (20/80) and ethyl acetate/hexane (30/70). Appropriate fractions were combined and concentrated to produce a white solid, which was then subjected to recrystallization. The product was dissolved in a minimal amount of hot methylene chloride, and hexane was added dropwise until cloudiness was observed. While cooling, needle-like crystals formed providing compound 4a (13.6 mg, 27%). mp 131–133 °C. 1H NMR (500 MHz, CDCl3) δ 7.51 (dd, J = 8.1, 2.4 Hz, 2H), 7.25 (d, J = 13.9 Hz, 1H), 7.21 (dd, J = 12.2, 7.2 Hz, 5H), 6.94 (d, J = 7.1 Hz, 2H), 6.75 (NH, d, J = 6.3 Hz, 1H), 4.96 (NH, d, J = 5.7 Hz, 1H), 3.86–3.80 (m, 1H), 3.39 (ddd, J = 14.3, 6.8, 2.4 Hz, 1H), 3.30–3.22 (m, 1H), 3.20 (OH, s, 1H), 2.95 (ttd, J = 14.1, 10.3, 8.5, 4.4 Hz, 2H), 2.43 (d, J = 2.4 Hz, 3H), 0.79 (s, 2H), 0.64–0.56 (m, 1H), 0.56–0.50 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 171.1, 144.2, 135.2, 135.0, 129.9, 129.1, 129.1, 127.4, 127.3, 58.2, 55.4, 47.7, 38.3, 21.6, 12.8, 12.5.
(S)-N-(2-hydroxy-2-methylpropyl)-2-((4-methylphenyl)sulfonamido)-3-phenylpropanamide (4b). Tosyl-L-phenylalanine (50 mg, 0.129 mmol) was reacted with dimethylethanolamine (14.4 µL, 0.155 mmol) following the general T3P coupling procedure and stirred at 50–60 °C for 6 days until completion. The crude product (oil) of 4b was crystallized from hot EA/hexane = 20/80. When the EA/hexane solution was cooled to rt, the product became an oil. Therefore, it is important to maintain the temperature of the solution to stay between 50–60 °C to allow a slow crystallization of the final product. Recrystallization provided compound 4b (29 mg, 58%) as clear needle-like crystals: mp 131–133 °C. 1H NMR (500 MHz, CDCl3) δ 7.53–7.47 (m, 2H), 7.23–7.14 (m, 5H), 6.96–6.90 (m, 2H), 6.74 (s, 1H), 4.93 (d, J = 6.2 Hz, 1H), 3.84 (dt, J = 7.8, 6.1 Hz, 1H), 3.29 (dd, J = 13.7, 6.9 Hz, 1H), 3.12 (dd, J = 13.7, 5.5 Hz, 1H), 2.97 (dd, J = 14.0, 5.9 Hz, 1H), 2.91 (dd, J = 14.0, 7.7 Hz, 1H), 2.43 (s, 3H), 1.16 (d, J = 2.2 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 170.9, 144.1, 135.2, 135.2, 129.9, 129.1, 129.0, 127.4, 127.2, 70.9, 58.1, 50.3, 38.3, 27.12, 27.1, 21.6. HRMS (ESI): Calcd for (MH+) C20H27N2O4S: 391.1686, found 391.1680.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

