

Differential Ability of Spike Protein of SARS-CoV-2 Variants to Downregulate ACE2
 , and
, and
Abstract
:1. Introduction
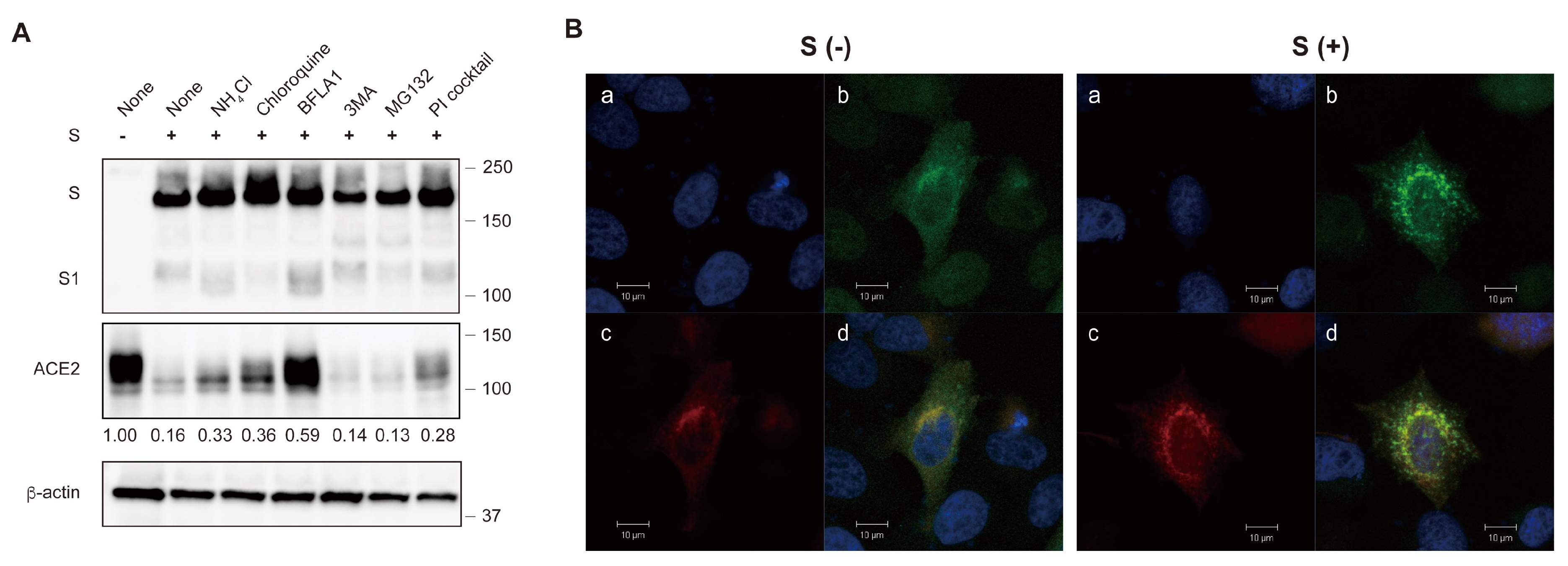
2. Results
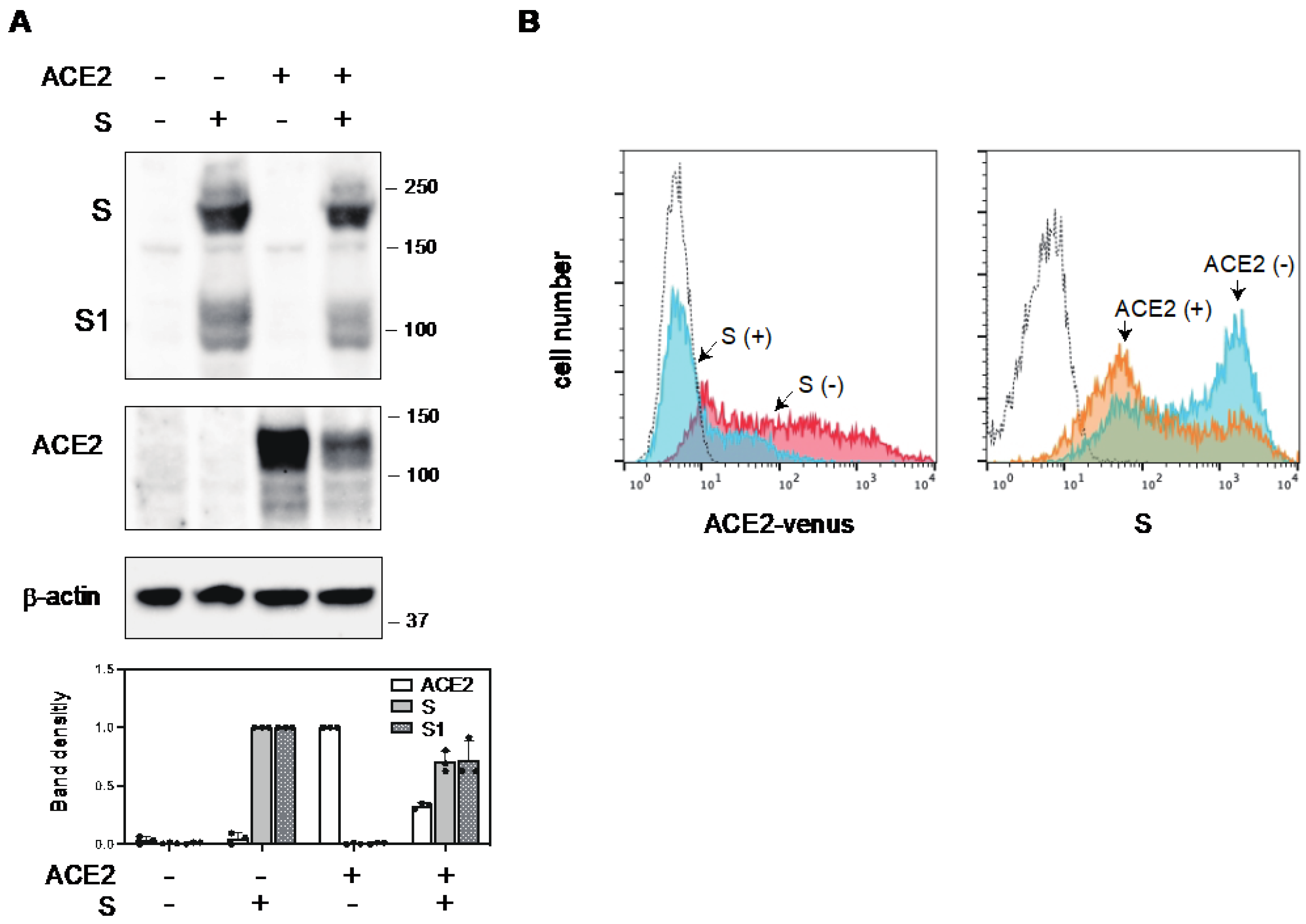
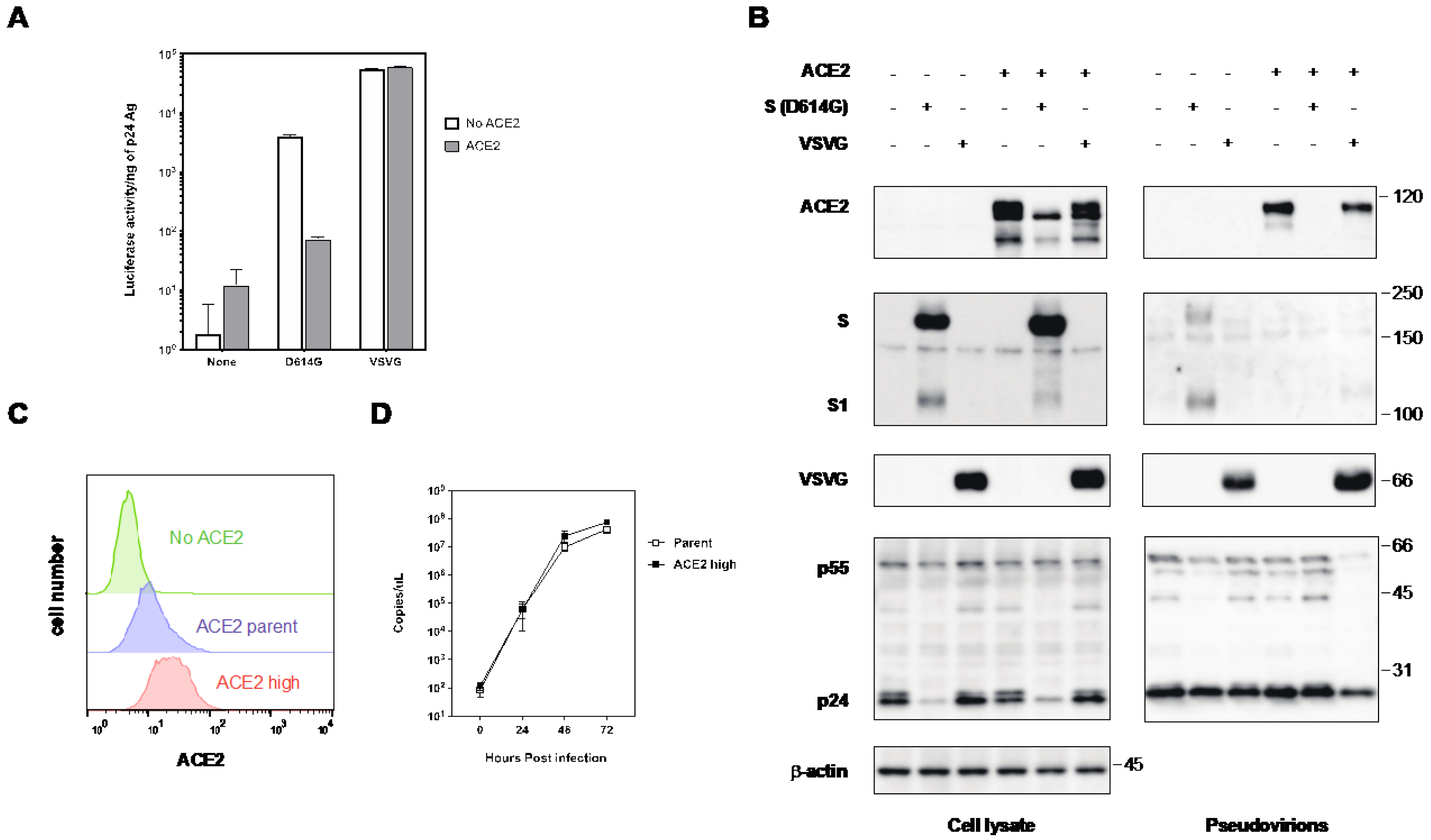
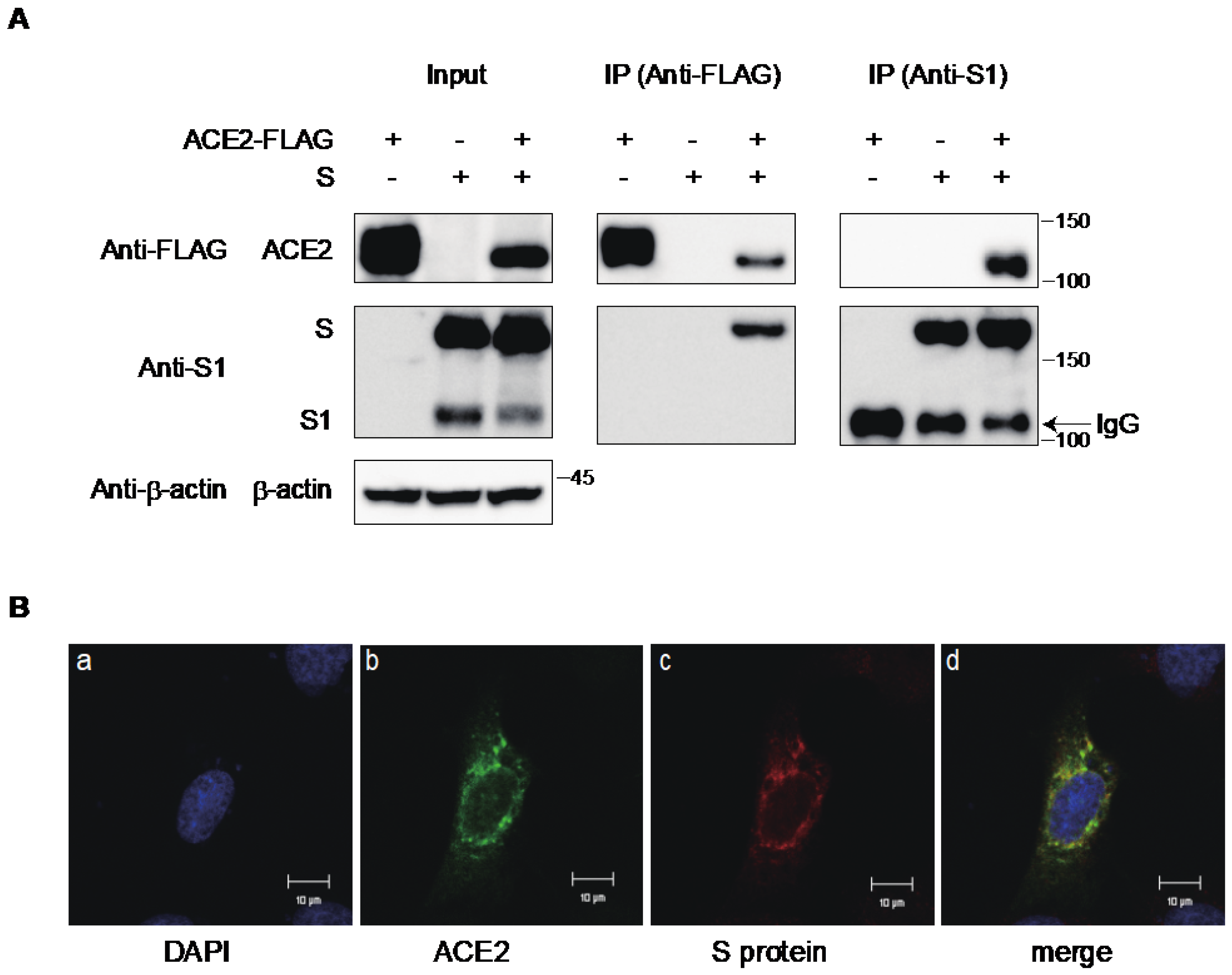
2.1. The S Protein of SARS-CoV-2 Downregulates ACE2
2.2. The Expression of ACE2 Impeded the Infectivity of Pseudovirus with the S Protein of SARS-CoV-2
2.3. Interaction of ACE2 with the S Protein of SARS-CoV-2 in the Cytoplasmic Compartment of the Cells
2.4. The S Protein Degrades ACE2 in the Lysosomal Compartment through the Endocytic Pathway
2.5. Cytoplasmic Domains of ACE2 and S Protein Are Not Essential for the Downregulation of ACE2
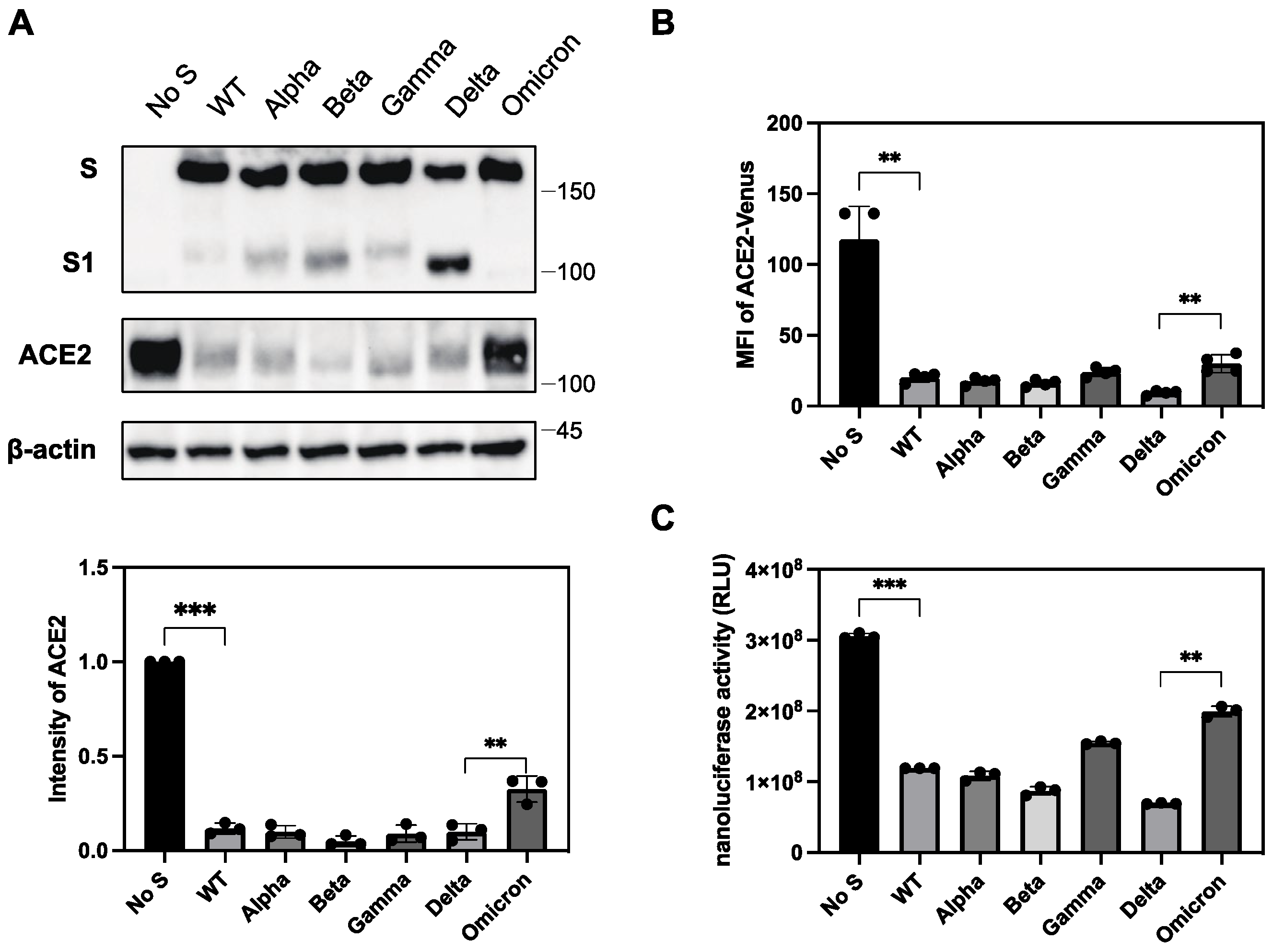
2.6. Differential Ability of S Protein from SARS-CoV-2 Variants to Downregulate ACE2
2.7. Determination of the Reduced ACE2 Downregulation Ability in Omicron Variants via the Receptor-Binding Domain and Heptad Repeat Domains of S the Protein
2.8. The Determination of the Increased ACE2 Downregulation Ability in the Delta Variants through the Combination of Three Substitutions in L452R/P681R/D950N of the S Protein
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents, and Viruses
4.2. Plasmids
4.3. Infection Experiments
4.4. Real-Time RT-PCR
4.5. Flow Cytometry
4.6. Quantitative Measurement of ACE2 Downregulation Activity by the S Protein from VOCs and Its Chimeras
4.7. Measurement of Fusion Activity of S Protein with ACE2
4.8. Immunoprecipitation and Western Blotting
4.9. Laser Scanning Confocal Microscopic Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): A review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 23 October 2023).
- WHO Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 23 October 2023).
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Bager, P.; Wohlfahrt, J.; Bhatt, S.; Stegger, M.; Legarth, R.; Moller, C.H.; Skov, R.L.; Valentiner-Branth, P.; Voldstedlund, M.; Fischer, T.K.; et al. Risk of hospitalisation associated with infection with SARS-CoV-2 omicron variant versus delta variant in Denmark: An observational cohort study. Lancet Infect. Dis. 2022, 22, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Mayr, F.B.; Talisa, V.B.; Castro, A.D.; Shaikh, O.S.; Omer, S.B.; Butt, A.A. COVID-19 disease severity in US Veterans infected during Omicron and Delta variant predominant periods. Nat. Commun. 2022, 13, 3647. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, T.; Ferguson, N.M.; Nash, S.G.; Webster, H.H.; Flaxman, S.; Andrews, N.; Hinsley, W.; Bernal, J.L.; Kall, M.; Bhatt, S.; et al. Comparative analysis of the risks of hospitalisation and death associated with SARS-CoV-2 omicron (B.1.1.529) and delta (B.1.617.2) variants in England: A cohort study. Lancet 2022, 399, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Sigal, A.; Milo, R.; Jassat, W. Estimating disease severity of Omicron and Delta SARS-CoV-2 infections. Nat. Rev. Immunol. 2022, 22, 267–269. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Silhol, F.; Sarlon, G.; Deharo, J.-C.; Vaïsse, B. Downregulation of ACE2 induces overstimulation of the renin–angiotensin system in COVID-19: Should we block the renin–angiotensin system? Hypertens. Res. 2020, 43, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Moniwa, N.; Takizawa, H.; Ura, N.; Shimamoto, K. Potential differential effects of renin-angiotensin system inhibitors on SARS-CoV-2 infection and lung injury in COVID-19. Hypertens. Res. 2020, 43, 837–840. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Chung, M.K.; Karnik, S.; Saef, J.; Bergmann, C.; Barnard, J.; Lederman, M.M.; Tilton, J.; Cheng, F.; Harding, C.V.; Young, J.B.; et al. SARS-CoV-2 and ACE2: The biology and clinical data settling the ARB and ACEI controversy. eBioMedicine 2020, 58, 102907. [Google Scholar] [CrossRef]
- Ramos, S.G.; Rattis, B.A.d.C.; Ottaviani, G.; Celes, M.R.N.; Dias, E.P. ACE2 down-regulation may act as a transient molecular disease causing RAAS dysregulation and tissue damage in the microcirculatory environment among COVID-19 patients. Am. J. Pathol. 2021, 191, 1154–1164. [Google Scholar] [CrossRef]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Levesque, K.; Zhao, Y.S.; Cohen, E.A. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J. Biol. Chem. 2003, 278, 28346–28353. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, C.A.; Tobiume, M.; Zhou, J.; Unutmaz, D.; Aiken, C. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J. Virol. 2002, 76, 4625–4633. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Terasawa, H.; Tanaka, Y.; Mitsuura, C.; Nakashima, K.; Yusa, K.; Harada, S. Separate cellular localizations of human T-lymphotropic virus 1 (HTLV-1) Env and glucose transporter type 1 (GLUT1) are required for HTLV-1 Env-mediated fusion and infection. J. Virol. 2015, 89, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Monde, K.; Maeda, Y.; Tanaka, Y.; Harada, S.; Yusa, K. Gp120 V3-dependent impairment of R5 HIV-1 infectivity due to virion-incorporated CCR5. J. Biol. Chem. 2007, 282, 36923–36932. [Google Scholar] [CrossRef]
- Moscona, A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005, 353, 1363–1373. [Google Scholar] [CrossRef]
- Tanaka, M.; Ueno, T.; Nakahara, T.; Sasaki, K.; Ishimoto, A.; Sakai, H. Downregulation of CD4 is required for maintenance of viral infectivity of HIV-1. Virology 2003, 311, 316–325. [Google Scholar] [CrossRef]
- Welstead, G.G.; Hsu, E.C.; Iorio, C.; Bolotin, S.; Richardson, C.D. Mechanism of CD150 (SLAM) down regulation from the host cell surface by measles virus hemagglutinin protein. J. Virol. 2004, 78, 9666–9674. [Google Scholar] [CrossRef]
- Wildum, S.; Schindler, M.; Münch, J.; Kirchhoff, F. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; tenOever, B.R.; Sanjana, N.E. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. eLife 2021, 10, e65365. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Kliche, J.; Kuss, H.; Ali, M.; Ivarsson, Y. Cytoplasmic short linear motifs in ACE2 and integrin β(3) link SARS-CoV-2 host cell receptors to mediators of endocytosis and autophagy. Sci. Signal 2021, 14, eabf1117. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.C.; Zhang, H.X.; Zhang, Z.; Rinkiko, S.; Cui, Y.M.; Zhu, Y.Z. The two-way switch role of ACE2 in the treatment of novel coronavirus pneumonia and underlying comorbidities. Molecules 2021, 26, 142. [Google Scholar] [CrossRef] [PubMed]
- Nasser, H.; Shimizu, R.; Ito, J.; Saito, A.; Sato, K.; Ikeda, T. Monitoring fusion kinetics of viral and target cell membranes in living cells using a SARS-CoV-2 spike-protein-mediated membrane fusion assay. STAR Protoc. 2022, 3, 101773. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Furusawa, Y.; Kiso, M.; Iida, S.; Uraki, R.; Hirata, Y.; Imai, M.; Suzuki, T.; Yamayoshi, S.; Kawaoka, Y. In SARS-CoV-2 delta variants, Spike-P681R and D950N promote membrane fusion, Spike-P681R enhances spike cleavage, but neither substitution affects pathogenicity in hamsters. eBioMedicine 2023, 91, 104561. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Sui, Y.; Li, J.; Venzon, D.J.; Berzofsky, J.A. SARS-CoV-2 Spike Protein Suppresses ACE2 and Type I Interferon Expression in Primary Cells From Macaque Lung Bronchoalveolar Lavage. Front. Immunol. 2021, 12, 658428. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, Q.; Fox, D.M.; Gao, C.; Stanley, S.A.; Luo, K. SARS-CoV-2 down-regulates ACE2 through lysosomal degradation. Mol. Biol. Cell 2022, 33, ar147. [Google Scholar] [CrossRef]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martínez-Sobrido, L.; García-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Klumperman, J.; Locker, J.K.; Meijer, A.; Horzinek, M.C.; Geuze, H.J.; Rottier, P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994, 68, 6523–6534. [Google Scholar] [CrossRef] [PubMed]
- Ujike, M.; Huang, C.; Shirato, K.; Makino, S.; Taguchi, F. The contribution of the cytoplasmic retrieval signal of severe acute respiratory syndrome coronavirus to intracellular accumulation of S proteins and incorporation of S protein into virus-like particles. J. Gen. Virol. 2016, 97, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Ogata, Y.; Mahiti, M.; Maeda, Y.; Kuang, X.T.; Miura, T.; Jessen, H.; Walker, B.D.; Brockman, M.A.; Brumme, Z.L.; et al. Differential ability of primary HIV-1 Nef isolates to downregulate HIV-1 entry receptors. J. Virol. 2015, 89, 9639–9652. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Nat. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Director-General’s Opening Remarks at the Media Briefing—5 May 2023. Available online: https://www.who.int/news-room/speeches/item/who-director-general-s-opening-remarks-at-the-media-briefing---5-may-2023 (accessed on 23 October 2023).
- Strasser, Z.H.; Greifer, N.; Hadavand, A.; Murphy, S.N.; Estiri, H. Estimates of SARS-CoV-2 Omicron BA.2 subvariant severity in New England. JAMA Netw. Open 2022, 5, e2238354. [Google Scholar] [CrossRef]
- Kaku, Y.; Kuwata, T.; Zahid, H.M.; Hashiguchi, T.; Noda, T.; Kuramoto, N.; Biswas, S.; Matsumoto, K.; Shimizu, M.; Kawanami, Y.; et al. Resistance of SARS-CoV-2 variants to neutralization by antibodies induced in convalescent patients with COVID-19. Cell Rep. 2021, 36, 109385. [Google Scholar] [CrossRef]
- Maeda, Y.; Foda, M.; Matsushita, S.; Harada, S. Involvement of both the V2 and V3 regions of the CCR5-tropic human immunodeficiency virus type 1 envelope in reduced sensitivity to macrophage inflammatory protein 1α. J. Virol. 2000, 74, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Takemura, T.; Chikata, T.; Kuwata, T.; Terasawa, H.; Fujimoto, R.; Kuse, N.; Akahoshi, T.; Murakoshi, H.; Tran, G.V.; et al. Existence of replication-competent minor variants with different coreceptor usage in plasma from HIV-1-infected individuals. J. Virol. 2020, 94, 1110–1128. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACE2 Downregulation Activity (%) a | Fusion Activity (%) b | |
|---|---|---|
| S wild | 100.0 ± 0.4 c | 100.0 ± 8.2 |
| Delta | 142.3 ± 0.5 | 257.7 ± 6.3 |
| Omicron | 75.4 ± 3.5 | 13.3 ± 1.6 |
| oddd | 134.8 ± 1.1 | 212.8 ± 8.6 |
| dodd | 93.5 ± 1.9 | 176.8 ± 5.8 |
| ddod | 113.3 ± 1.2 | 95.6 ± 4.8 |
| dddo | 110.2 ± 2.7 | 127.9 ± 5.2 |
| oodd | 111.7 ± 3.3 | 184.9 ± 9.0 |
| odod | 104.1 ± 2.4 | 71.1 ± 5.8 |
| oddo | 107.0 ± 3.7 | 117.3 ± 3.9 |
| dood | 104.6 ± 2.2 | 83.7 ± 9.8 |
| dodo | 47.8 ± 3.9 | 48.4 ± 2.6 |
| ddoo | 88.5 ± 3.1 | 26.2 ± 0.7 |
| oood | 117.4 ± 3.0 | 67.5 ± 3.7 |
| oodo | 67.7 ± 1.2 | 46.7 ± 4.7 |
| odoo | 77.5 ± 2.0 | 24.5 ± 3.1 |
| dooo | 75.9 ± 2.2 | 13.4 ± 1.1 |
| ACE2 Downregulation Activity (%) a | Fusion Activity (%) b | |
|---|---|---|
| S wild (Wuhan) | 100.0 ± 5.9 c | 100.0 ± 5.7 c |
| Delta | 156.4 ± 4.8 | 196.5 ± 18.3 |
| Omicron | 71.0 ± 7.7 | 14.3 ± 2.6 |
| L452R | 107.6 ± 5.5 | 97.0 ± 9.3 |
| P681R | 95.3 ± 11.4 | 166.5 ± 12.1 |
| D950N | 117.1 ± 4.4 | 124.2 ± 10.2 |
| L452R/P681R | 120.6 ± 6.4 | 170.8 ± 12.9 |
| L452R/D950N | 111.8 ± 5.7 | 122.2 ± 5.2 |
| P681R/D950N | 133.5 ± 4.8 | 196.6 ± 10.7 |
| L452R/P681R/D950N | 156.6 ± 5.9 | 187.5 ± 6.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, Y.; Toyoda, M.; Kuwata, T.; Terasawa, H.; Tokugawa, U.; Monde, K.; Sawa, T.; Ueno, T.; Matsushita, S. Differential Ability of Spike Protein of SARS-CoV-2 Variants to Downregulate ACE2. Int. J. Mol. Sci. 2024, 25, 1353. https://doi.org/10.3390/ijms25021353
Maeda Y, Toyoda M, Kuwata T, Terasawa H, Tokugawa U, Monde K, Sawa T, Ueno T, Matsushita S. Differential Ability of Spike Protein of SARS-CoV-2 Variants to Downregulate ACE2. International Journal of Molecular Sciences. 2024; 25(2):1353. https://doi.org/10.3390/ijms25021353
Chicago/Turabian StyleMaeda, Yosuke, Mako Toyoda, Takeo Kuwata, Hiromi Terasawa, Umiru Tokugawa, Kazuaki Monde, Tomohiro Sawa, Takamasa Ueno, and Shuzo Matsushita. 2024. "Differential Ability of Spike Protein of SARS-CoV-2 Variants to Downregulate ACE2" International Journal of Molecular Sciences 25, no. 2: 1353. https://doi.org/10.3390/ijms25021353
APA StyleMaeda, Y., Toyoda, M., Kuwata, T., Terasawa, H., Tokugawa, U., Monde, K., Sawa, T., Ueno, T., & Matsushita, S. (2024). Differential Ability of Spike Protein of SARS-CoV-2 Variants to Downregulate ACE2. International Journal of Molecular Sciences, 25(2), 1353. https://doi.org/10.3390/ijms25021353