
The Association of Hippocampal Long-Term Potentiation-Induced Gene Expression with Genetic Risk for Psychosis

 ,
,
Abstract
:1. Introduction
2. Results
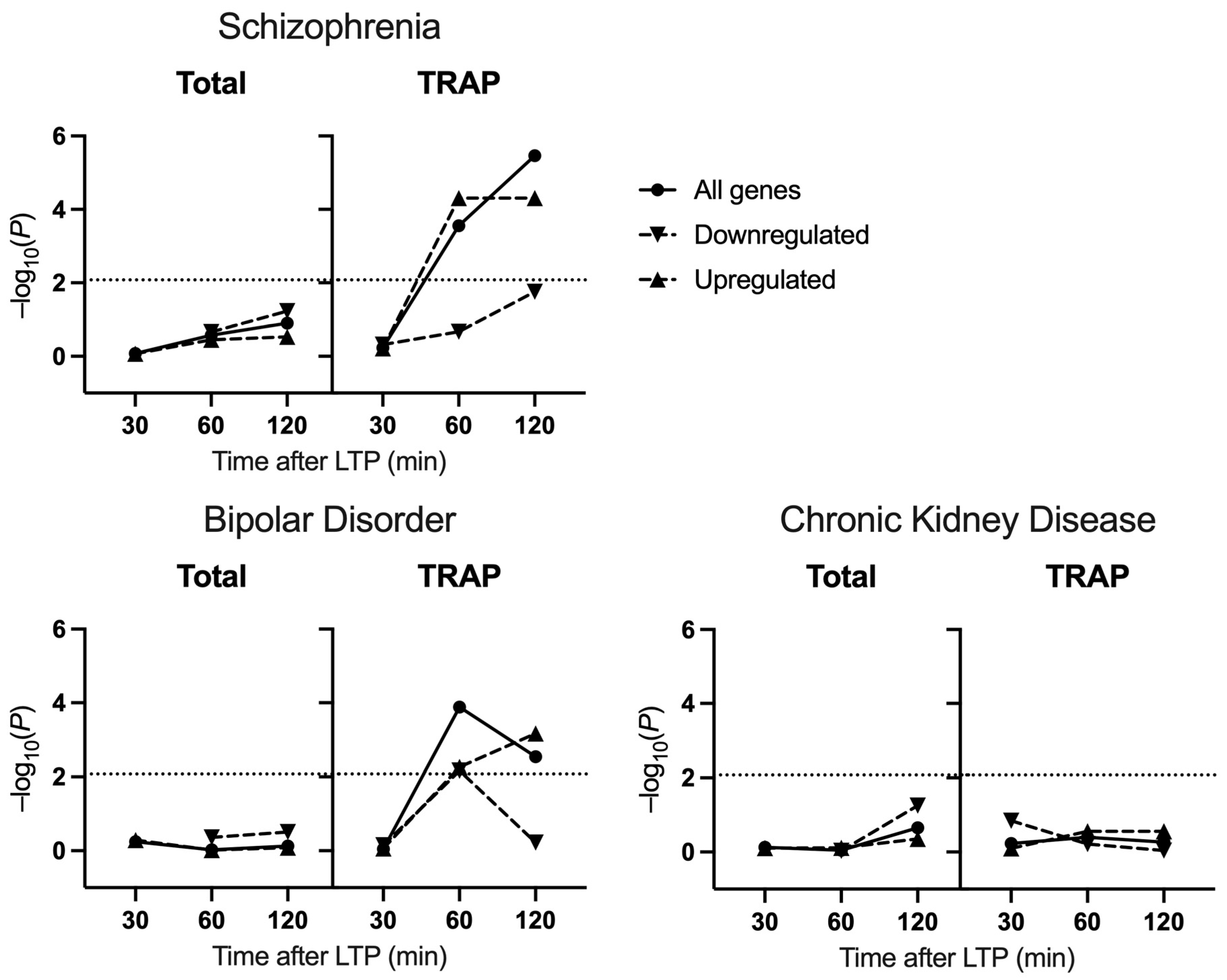
2.1. Common Variant Association
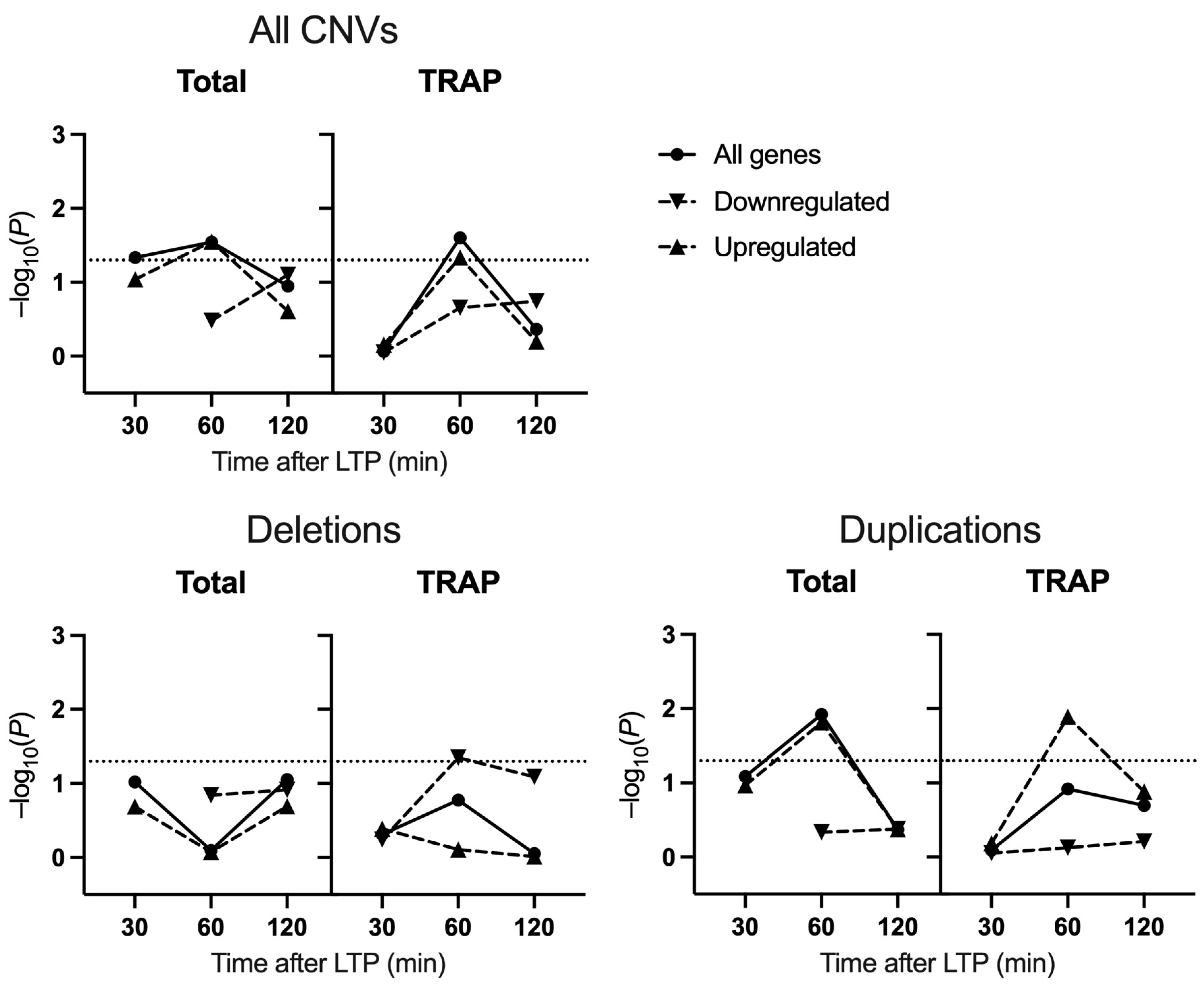
2.2. Schizophrenia CNV Enrichment
2.3. Schizophrenia De Novo Rare Coding Variant Enrichment
3. Discussion
4. Materials and Methods
4.1. LTP-Induced Gene Sets
4.2. Genotype Data
4.3. Gene Set Enrichment Analysis
probes + CNV study + number of gene set hits
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, J.; Bray, N.J. Schizophrenia genomics: Convergence on synaptic development, adult synaptic plasticity, or both? Biol. Psychiatry 2022, 91, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Mould, A.W.; Hall, N.A.; Milosevic, I.; Tunbridge, E.M. Targeting synaptic plasticity in schizophrenia: Insights from genomic studies. Trends Mol. Med. 2021, 27, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Green, T.; Gothelf, D.; Glaser, B.; Debbane, M.; Frisch, A.; Kotler, M.; Weizman, A.; Eliez, S. Psychiatric Disorders and Intellectual Functioning Throughout Development in Velocardiofacial (22q11.2 Deletion) Syndrome. J. Am. Acad. Child Adolesc. Psychiatry 2009, 48, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Howrigan, D.P.; Merico, D.; Thiruvahindrapuram, B.; Wu, W.; Greer, D.S.; Antaki, D.; Shetty, A.; Holmans, P.A.; Pinto, D.; et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 2017, 49, 27–35. [Google Scholar] [CrossRef]
- Rees, E.; Walters, J.T.R.; Georgieva, L.; Isles, A.R.; Chambert, K.D.; Richards, A.L.; Mahoney-Davies, G.; Legge, S.E.; Moran, J.L.; McCarroll, S.A.; et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br. J. Psychiatry 2014, 204, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.P.H.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buiz er-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef]
- Rees, E.; Kendall, K.; Pardiñas, A.F.; Legge, S.E.; Pocklington, A.; Escott-Price, V.; MacCabe, J.H.; Collier, D.A.; Holmans, P.; O’donovan, M.C.; et al. Analysis of Intellectual Disability Copy Number Variants for Association With Schizophrenia. JAMA Psychiatry 2016, 73, 963–969. [Google Scholar] [CrossRef]
- Rees, E.; Carrera, N.; Morgan, J.; Hambridge, K.; Escott-Price, V.; Pocklington, A.J.; Richards, A.L.; Pardiñas, A.F.; McDonald, C.; Donohoe, G.; et al. Targeted Sequencing of 10,198 Samples Confirms Abnormalities in Neuronal Activity and Implicates Voltage-Gated Sodium Channels in Schizophrenia Pathogenesis. Biol. Psychiatry 2019, 85, 554–562. [Google Scholar] [CrossRef]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022, 604, 509–516. [Google Scholar] [CrossRef]
- Rees, E.; Creeth, H.D.J.; Hwu, H.-G.; Chen, W.J.; Tsuang, M.; Glatt, S.J.; Rey, R.; Kirov, G.; Walters, J.T.R.; Holmans, P.; et al. Schizophrenia, autism spectrum disorders and developmental disorders share specific disruptive coding mutations. Nat. Commun. 2021, 12, 5353. [Google Scholar] [CrossRef]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Pocklington, A.J.; Rees, E.; Walters, J.T.R.; Han, J.; Kavanagh, D.H.; Chambert, K.D.; Holmans, P.; Moran, J.L.; McCarroll, S.A.; Kirov, G.; et al. Novel Findings from CNVs Implicate Inhibitory and Excitatory Signaling Complexes in Schizophrenia. Neuron 2015, 86, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.A.; Breen, G.; Forstner, A.J.; McQuillin, A.; Ripke, S.; Trubetskoy, V.; Mattheisen, M.; Wang, Y.; Coleman, J.R.I.; Gaspar, H.A.; et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019, 51, 793–803. [Google Scholar] [CrossRef]
- Mullins, N.; Forstner, A.J.; O’connell, K.S.; Coombes, B.; Coleman, J.R.I.; Qiao, Z.; Als, T.D.; Bigdeli, T.B.; Børte, S.; Bryois, J.; et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 2021, 53, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef]
- Bliss, T.V.P.; Lømo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Whitlock, J.R.; Heynen, A.J.; Shuler, M.G.; Bear, M.F. Learning Induces Long-Term Potentiation in the Hippocampus. Science 2006, 313, 1093–1097. [Google Scholar] [CrossRef]
- Frey, U.; Krug, M.; Reymann, K.G.; Matthies, H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988, 452, 57–65. [Google Scholar] [CrossRef]
- Bradshaw, K.D.; Emptage, N.J.; Bliss, T.V.P. A role for dendritic protein synthesis in hippocampal late LTP. Eur. J. Neurosci. 2003, 18, 3150–3152. [Google Scholar] [CrossRef]
- Redondo, R.L.; Morris, R.G.M. Making memories last: The synaptic tagging and capture hypothesis. Nat. Rev. Neurosci. 2011, 12, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Asok, A.; Leroy, F.; Rayman, J.B.; Kandel, E.R. Molecular Mechanisms of the Memory Trace. Trends Neurosci. 2019, 42, 14–22. [Google Scholar] [CrossRef]
- Chen, P.B.; Kawaguchi, R.; Blum, C.; Achiro, J.M.; Coppola, G.; O’Dell, T.J.; Martin, K.C. Mapping Gene Expression in Excitatory Neurons during Hippocampal Late-Phase Long-Term Potentiation. Front. Mol. Neurosci. 2017, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Orfila, J.E.; Achanta, P.; Martinez, J.L. Gene expression associated with in vivo induction of early phase-long-term potentiation (LTP) in the hippocampal mossy fiber-Cornus Ammonis (CA)3 pathway. Cell. Mol. Biol. 2003, 49, 1281–1287. [Google Scholar]
- Wibrand, K.; Messaoudi, E.; Håvik, B.; Steenslid, V.; Løvlie, R.; Steen, V.M.; Bramham, C.R. Identification of genes co-upregulated with Arc during BDNF-induced long-term potentiation in adult rat dentate gyrus in vivo. Eur. J. Neurosci. 2006, 23, 1501–1511. [Google Scholar] [CrossRef]
- Valor, L.M.; Barco, A. Hippocampal gene profiling: Toward a systems biology of the hippocampus. Hippocampus 2012, 22, 929–941. [Google Scholar] [CrossRef]
- Ryan, M.M.; Ryan, B.; Kyrke-Smith, M.; Logan, B.; Tate, W.P.; Abraham, W.C.; Williams, J.M. Temporal Profiling of Gene Networks Associated with the Late Phase of Long-Term Potentiation In Vivo. PLoS ONE 2012, 7, e40538. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Gong, R.; Stuart, J.; Tang, S.-J. Molecular Network and Chromosomal Clustering of Genes Involved in Synaptic Plasticity in the Hippocampus. J. Biol. Chem. 2006, 281, 30195–30211. [Google Scholar] [CrossRef]
- Doyle, J.P.; Dougherty, J.D.; Heiman, M.; Schmidt, E.F.; Stevens, T.R.; Ma, G.; Bupp, S.; Shrestha, P.; Shah, R.D.; Doughty, M.L.; et al. Application of a Translational Profiling Approach for the Comparative Analysis of CNS Cell Types. Cell 2008, 135, 749–762. [Google Scholar] [CrossRef]
- Haimon, Z.; Volaski, A.; Orthgiess, J.; Boura-Halfon, S.; Varol, D.; Shemer, A.; Yona, S.; Zuckerman, B.; David, E.; Chappell-Maor, L.; et al. Re-evaluating microglia expression profiles using RiboTag and cell isolation strategies. Nat. Immunol. 2018, 19, 636–644. [Google Scholar] [CrossRef]
- Sanz, E.; Bean, J.C.; Carey, D.P.; Quintana, A.; McKnight, G.S. RiboTag: Ribosomal Tagging Strategy to Analyze Cell-Type-Specific mRNA Expression In Vivo. Curr. Protoc. Neurosci. 2019, 88, e77. [Google Scholar] [CrossRef] [PubMed]
- Tsien, J.Z.; Chen, D.F.; Gerber, D.; Tom, C.; Mercer, E.H.; Anderson, D.J.; Mayford, M.; Kandel, E.R.; Tonegawa, S. Subregion- and Cell Type–Restricted Gene Knockout in Mouse Brain. Cell 1996, 87, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Skene, N.G.; Bryois, J.; Bakken, T.E.; Breen, G.; Crowley, J.J.; Gaspar, H.A.; Giusti-Rodriguez, P.; Hodge, R.D.; Miller, J.A.; Muñoz-Manchado, A.B.; et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018, 50, 825–833. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Alberini, C.M.; Kandel, E.R. The Regulation of Transcription in Memory Consolidation. Cold Spring Harb. Perspect. Biol. 2014, 7, a021741. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.-J. Structural LTP: Signal transduction, actin cytoskeleton reorganization, and membrane remodeling of dendritic spines. Curr. Opin. Neurobiol. 2022, 74, 102534. [Google Scholar] [CrossRef]
- Lee, P.H.; Feng, Y.-C.A.; Smoller, J.W. Pleiotropy and Cross-Disorder Genetics Among Psychiatric Disorders. Biol. Psychiatry 2021, 89, 20–31. [Google Scholar] [CrossRef]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef]
- Pembroke, W.G.; Hartl, C.L.; Geschwind, D.H. Evolutionary conservation and divergence of the human brain transcriptome. Genome Biol. 2021, 22, 52. [Google Scholar] [CrossRef]
- Emes, R.D.; Grant, S.G.N. Evolution of Synapse Complexity and Diversity. Annu. Rev. Neurosci. 2012, 35, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Gobert, D.; Topolnik, L.; Azzi, M.; Huang, L.; Badeaux, F.; Desgroseillers, L.; Sossin, W.S.; Lacaille, J.-C. Forskolin induction of late-LTP and up-regulation of 5′ TOP mRNAs translation via mTOR, ERK, and PI3K in hippocampal pyramidal cells. J. Neurochem. 2008, 106, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Otmakhov, N.; Khibnik, L.; Otmakhova, N.; Carpenter, S.; Riahi, S.; Asrican, B.; Lisman, J. Forskolin-Induced LTP in the CA1 Hippocampal Region Is NMDA Receptor Dependent. J. Neurophysiol. 2004, 91, 1955–1962. [Google Scholar] [CrossRef]
- Bigdeli, T.B.; Genovese, G.; Georgakopoulos, P.; Meyers, J.L.; Peterson, R.E.; Iyegbe, C.O.; Medeiros, H.; Valderrama, J.; Achtyes, E.D.; Kotov, R.; et al. Contributions of common genetic variants to risk of schizophrenia among individuals of African and Latino ancestry. Mol. Psychiatry 2020, 25, 2455–2467. [Google Scholar] [CrossRef]
- Wuttke, M.; Li, Y.; Li, M.; Sieber, K.B.; Feitosa, M.F.; Gorski, M.; Tin, A.; Wang, L.; Chu, A.Y.; Hoppmann, A.; et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 2020, 51, 957–972. [Google Scholar] [CrossRef]
- Rees, E.; Han, J.; Morgan, J.; Carrera, N.; Escott-Price, V.; Pocklington, A.J.; Duffield, M.; Hall, L.S.; Legge, S.E.; Pardiñas, F.; et al. De novo mutations identified by exome sequencing implicate rare missense variants in SLC6A1 in schizophrenia. Nat. Neurosci. 2020, 23, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ambalavanan, A.; Girard, S.L.; Ahn, K.; Zhou, S.; Dionne-Laporte, A.; Spiegelman, D.; Bourassa, C.V.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; et al. De novo variants in sporadic cases of childhood onset schizophrenia. Eur. J. Hum. Genet. 2016, 24, 944–948. [Google Scholar] [CrossRef]
- Girard, S.L.; Gauthier, J.; Noreau, A.; Xiong, L.; Zhou, S.; Jouan, L.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, E.; Diallo, O.; et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011, 43, 860–863. [Google Scholar] [CrossRef]
- Guipponi, M.; Santoni, F.; Setola, V.; Gehrig, C.; Rotharmel, M.; Cuenca, M.; Guillin, O.; Dikeos, D.; Georgantopoulos, G.; Papadimitriou, G.; et al. Exome Sequencing in 53 Sporadic Cases of Schizophrenia Identifies 18 Putative Candidate Genes. PLoS ONE 2014, 9, e112745. [Google Scholar] [CrossRef]
- Gulsuner, S.; Walsh, T.; Watts, A.C.; Lee, M.K.; Thornton, A.M.; Casadei, S.; Rippey, C.; Shahin, H.; Nimgaonkar, V.L.; Go, R.C.; et al. Spatial and Temporal Mapping of De Novo Mutations in Schizophrenia to a Fetal Prefrontal Cortical Network. Cell 2013, 154, 518–529. [Google Scholar] [CrossRef]
- E McCarthy, S.; Gillis, J.; Kramer, M.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef]
- Takata, A.; Xu, B.; Ionita-Laza, I.; Roos, J.L.; Gogos, J.A.; Karayiorgou, M. Loss-of-Function Variants in Schizophrenia Risk and SETD1A as a Candidate Susceptibility Gene. Neuron 2014, 82, 773–780. [Google Scholar] [CrossRef]
- Wang, Q.; Li, M.; Yang, Z.; Hu, X.; Wu, H.-M.; Ni, P.; Ren, H.; Deng, W.; Li, M.; Ma, X.; et al. Increased co-expression of genes harboring the damaging de novo mutations in Chinese schizophrenic patients during prenatal development. Sci. Rep. 2015, 5, 18209. [Google Scholar] [CrossRef]
- Xu, B.; Ionita-Laza, I.; Roos, J.L.; Boone, B.; Woodrick, S.; Sun, Y.; Levy, S.; Gogos, J.A.; Karayiorgou, M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 2012, 44, 1365–1369. [Google Scholar] [CrossRef]
- Samocha, K.E.; Kosmicki, J.A.; Karczewski, K.J.; O’Donnell-Luria, A.H.; Pierce-Hoffman, E.; MacArthur, D.G.; Neale, B.M.; Daly, M.J. Regional missense constraint improves variant deleteriousness prediction. BioRxiv 2017. Preprint. [Google Scholar] [CrossRef]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008, 455, 237–241. [Google Scholar] [CrossRef]
- Levinson, D.F.; Duan, J.; Oh, S.; Wang, K.; Sanders, A.R.; Shi, J.; Zhang, N.; Mowry, B.J.; Olincy, A.; Amin, F.; et al. Copy Number Variants in Schizophrenia: Confirmation of Five Previous Findings and New Evidence for 3q29 Microdeletions and VIPR2 Duplications. Am. J. Psychiatry 2011, 168, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Samocha, K.E.; Robinson, E.B.; Sanders, S.J.; Stevens, C.; Sabo, A.; McGrath, L.M.; Kosmicki, J.A.; Rehnström, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Bipolar Disorder Subtype | β Value (SE) | p-Value | Adjusted p-Value | |
|---|---|---|---|---|
| 30 min TRAP-seq | Bipolar I | −0.090 (0.13) | 0.76 | 1.0 |
| Bipolar II | −0.090 (0.11) | 0.80 | 1.0 | |
| 30 min total RNA-seq | Bipolar I | −0.066 (0.22) | 0.62 | 1.0 |
| Bipolar II | −0.11 (0.18) | 0.72 | 1.0 | |
| 60 min TRAP-seq | Bipolar I | 0.13 (0.029) | 3.1 × 10−6 | 1.9 × 10−5 |
| Bipolar II | 0.0025 (0.025) | 0.46 | 1.0 | |
| 60 min total RNA-seq | Bipolar I | −0.027 (0.10) | 0.60 | 0.898 |
| Bipolar II | −0.096 (0.091) | 0.85 | 1.0 | |
| 120 min TRAP-seq | Bipolar I | 0.094 (0.034) | 0.0027 | 0.016 |
| Bipolar II | 0.0070 (0.029) | 0.40 | 1.0 | |
| 120 min total RNA-seq | Bipolar I | −0.028 (0.050) | 0.71 | 1.0 |
| Bipolar II | −0.028 (0.043) | 0.74 | 1.0 |
| LTP Gene Set | Schizophrenia Ultra-Rare Coding Variant Enrichment | |||
|---|---|---|---|---|
| Observed/Expected | p-Value | Adjusted p-Value | Rate Ratio (95% Confidence Intervals) | |
| 30-min total | 2/0.497 | 0.116 | 0.736 | 3.45 (0.416–12.5) |
| 30-min TRAP | 5/3.45 | 0.610 | 1.00 | 1.24 (0.401–2.91) |
| 60-min total | 7/3.27 | 0.114 | 0.642 | 1.84 (0.737–3.82) |
| 60-min TRAP | 80/57.4 | 0.101 | 0.931 | 1.23 (0.954–1.56) |
| 120-min total | 17/14.2 | 0.900 | 1.00 | 1.02 (0.592–1.66) |
| 120-min TRAP | 80/51.4 | 0.00892 | 0.181 | 1.39 (1.08–1.77) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wellard, N.L.; Clifton, N.E.; Rees, E.; Thomas, K.L.; Hall, J. The Association of Hippocampal Long-Term Potentiation-Induced Gene Expression with Genetic Risk for Psychosis. Int. J. Mol. Sci. 2024, 25, 946. https://doi.org/10.3390/ijms25020946
Wellard NL, Clifton NE, Rees E, Thomas KL, Hall J. The Association of Hippocampal Long-Term Potentiation-Induced Gene Expression with Genetic Risk for Psychosis. International Journal of Molecular Sciences. 2024; 25(2):946. https://doi.org/10.3390/ijms25020946
Chicago/Turabian StyleWellard, Natalie L., Nicholas E. Clifton, Elliott Rees, Kerrie L. Thomas, and Jeremy Hall. 2024. "The Association of Hippocampal Long-Term Potentiation-Induced Gene Expression with Genetic Risk for Psychosis" International Journal of Molecular Sciences 25, no. 2: 946. https://doi.org/10.3390/ijms25020946
APA StyleWellard, N. L., Clifton, N. E., Rees, E., Thomas, K. L., & Hall, J. (2024). The Association of Hippocampal Long-Term Potentiation-Induced Gene Expression with Genetic Risk for Psychosis. International Journal of Molecular Sciences, 25(2), 946. https://doi.org/10.3390/ijms25020946