

Targeting Innate Immunity in Glioma Therapy

 , ,
, ,
Abstract
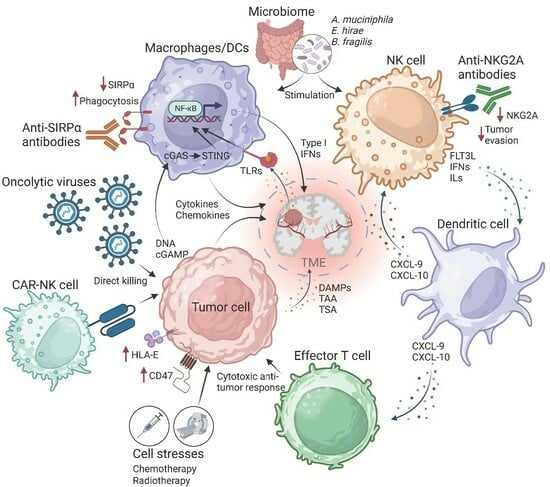
:
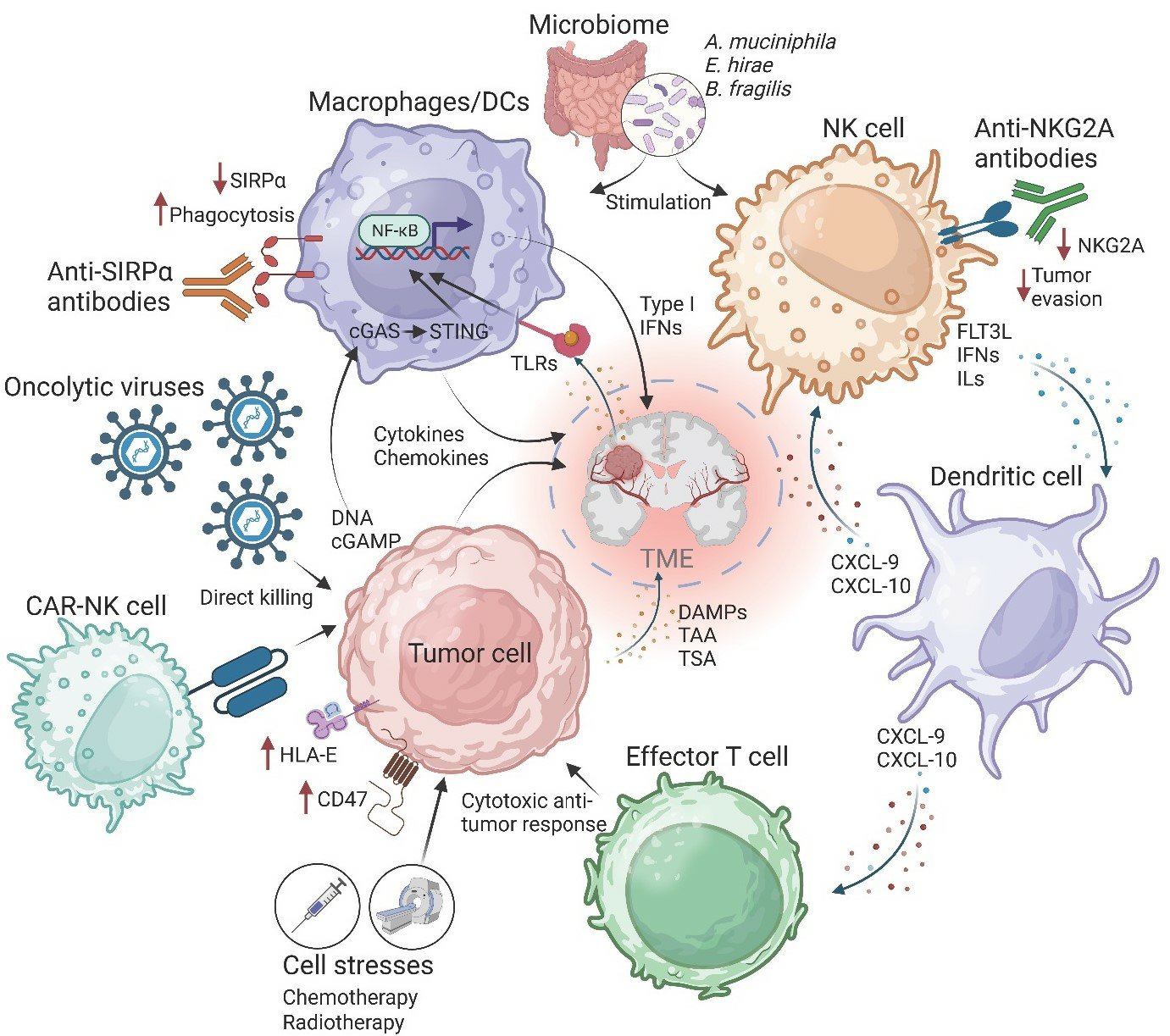{kind=link}
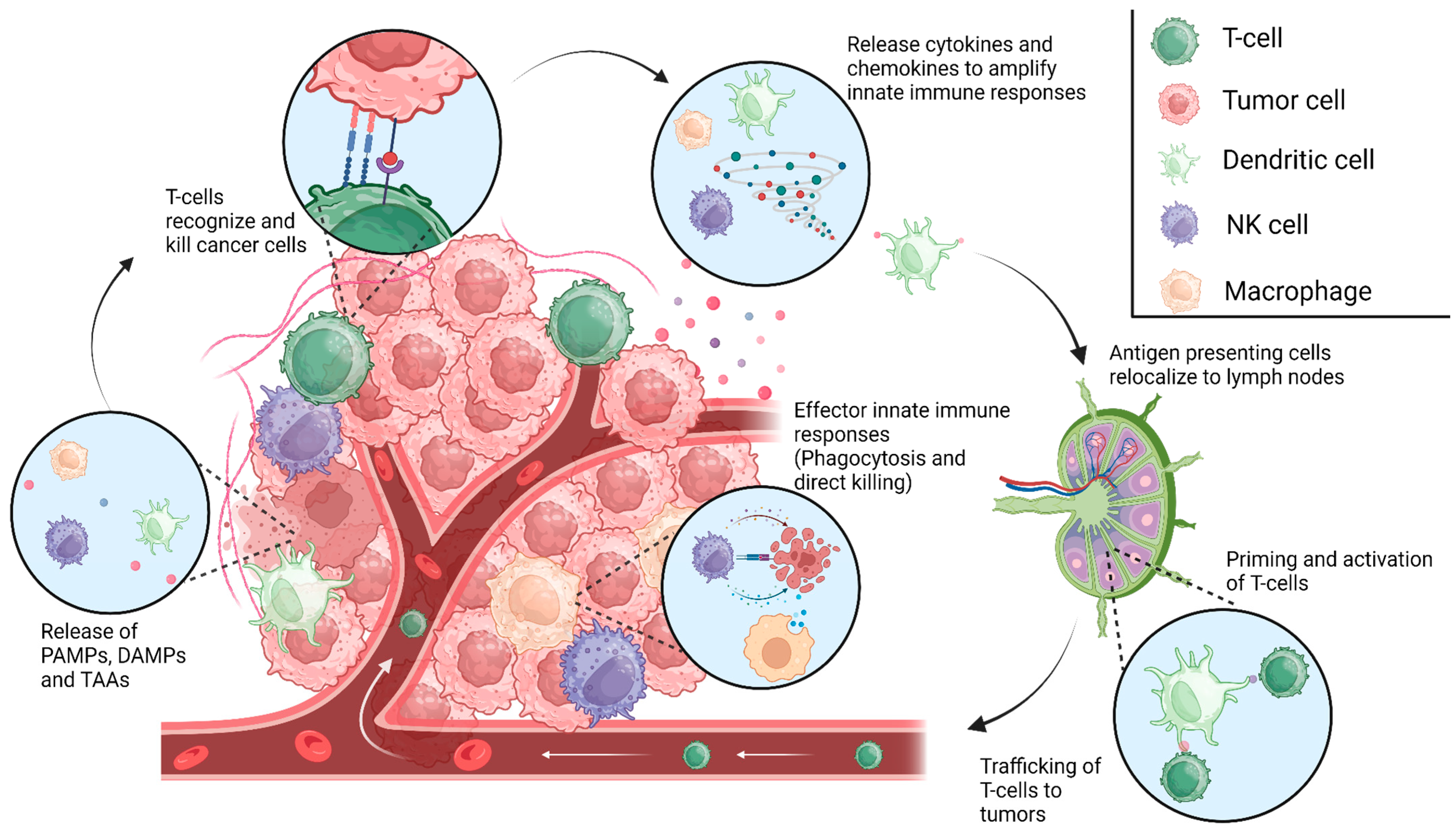{kind=link}
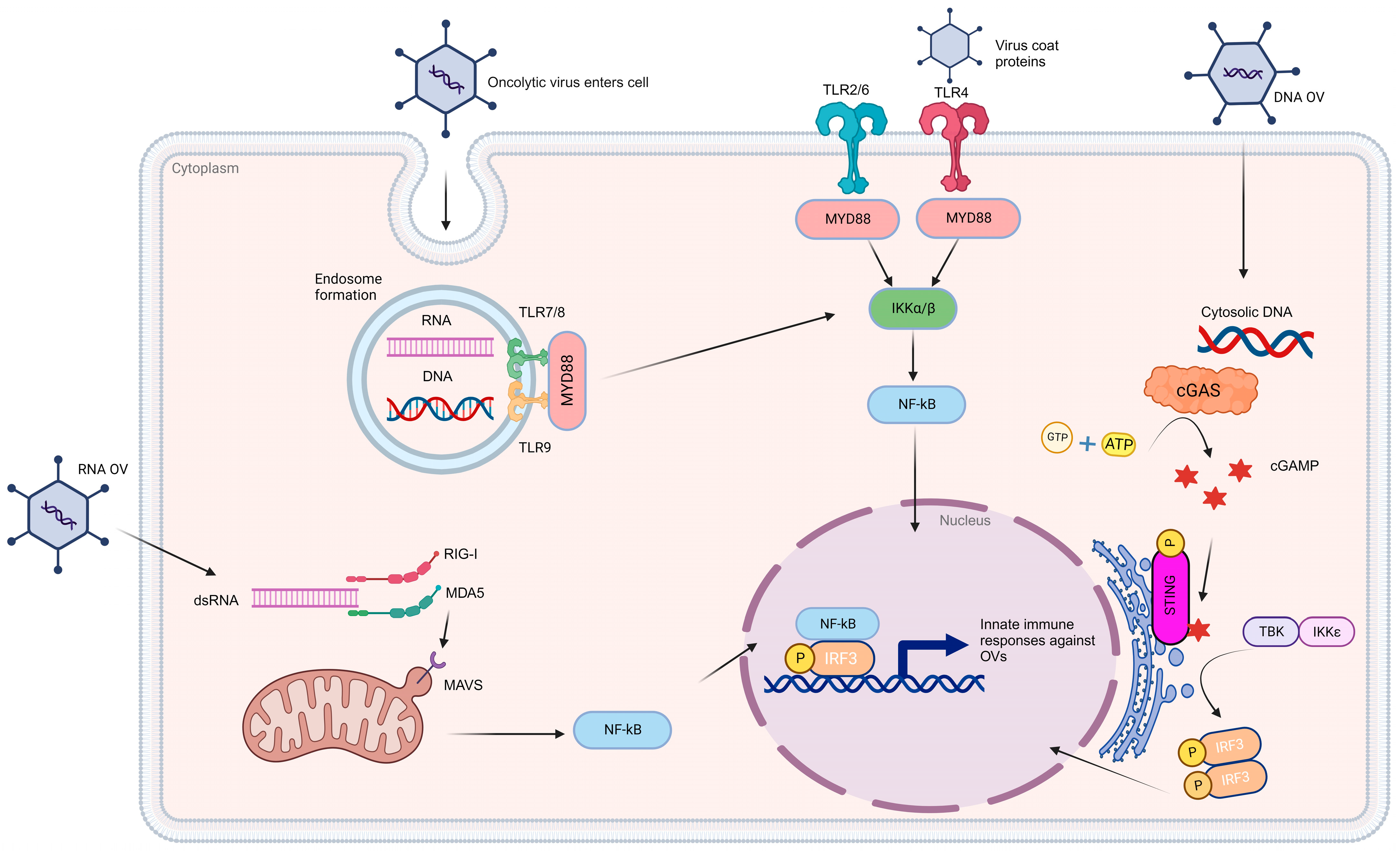{kind=link}
1. Introduction
2. Discussion
2.1. Activation of Toll-like Receptors
2.2. Treatments That Induce a Stress Response
2.3. Strategies to Amplify the Innate Immune Response
2.4. Type I Interferons
2.5. Direct and Indirect Activation of Innate Immunity Using Therapeutic Antibodies
2.6. Innate Immune Checkpoint Therapy
2.7. CAR-NK Cells
2.8. Oncolytic Virotherapy
2.9. Manipulation of Microbiome and Innate Immunity against Cancer
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro-Oncology 2023, 25, IV1–IV99. [Google Scholar] [CrossRef]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. J. Am. Med. Assoc. 2023, 329, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Villa, S.; Manes, A.; Hostalot, C.; Capellades, J.; Costas, E.; Rosinol, O.; Sanz, C.; Rosell, R.; Balañà, C. Short course of radiotherapy and concomitant temozolomide in patients affected with glioblastoma with V–VI prognostic classes: A pilot study. Neuro-Oncology 2009, 11, 966–967. [Google Scholar]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Lim, M. Advancing combination therapy for recurrent glioblastoma. Nat. Med. 2023, 29, 1318–1319. [Google Scholar] [CrossRef] [PubMed]
- See, A.P.; Parker, J.J.; Waziri, A. The role of regulatory T cells and microglia in glioblastoma-associated immunosuppression. J. Neuro-Oncol. 2015, 123, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Cao, L.L.; Kagan, J.C. Targeting innate immune pathways for cancer immunotherapy. Immunity 2023, 56, 2206–2217. [Google Scholar] [CrossRef]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Publisher Correction: Harnessing innate immunity in cancer therapy. Nature 2019, 576, E3. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Pasare, C.; Medzhitov, R. Toll-like receptors: Linking innate and adaptive immunity. Microbes Infect. 2004, 6, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xie, Y.; Sun, X.; Zeh, H.J.; Kang, R.; Lotze, M.T.; Tang, D. DAMPs, ageing, and cancer: The ‘DAMP Hypothesis’. Ageing Res. Rev. 2015, 24, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017, 27, 96–108. [Google Scholar] [CrossRef]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.-H.; Pagès, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef]
- Bogolyubova, A.V.; Belousov, P.V. Inflammatory immune infiltration in human tumors: Role in pathogenesis and prognostic and diagnostic value. Biochemistry 2016, 81, 1261–1273. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Lu, M.; Wen, P.Y.; Taylor, J.W.; Maher, E.A.; Arrillaga-Romany, I.; Peters, K.B.; Ellingson, B.M.; Rosenblum, M.K.; Chun, S.; et al. Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: A randomized, perioperative phase 1 trial. Nat. Med. 2023, 29, 615–622. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A. Innate immune induction of the adaptive immune response. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.-X.; Xu, M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef]
- Wargo, J.A.; Cooper, Z.A.; Flaherty, K.T. Universes collide: Combining immunotherapy with targeted therapy for cancer. Cancer Discov. 2014, 4, 1377–1386. [Google Scholar] [CrossRef]
- Simone, C.B.; Burri, S.H.; Heinzerling, J.H. Novel radiotherapy approaches for lung cancer: Combining radiation therapy with targeted and immunotherapies. Transl. Lung Cancer Res. 2015, 4, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Cen, X.; Liu, S.; Cheng, K. The role of toll-like receptor in inflammation and tumor immunity. Front. Pharmacol. 2018, 9, 878. [Google Scholar] [CrossRef]
- Clarke, S.R.M. The critical role of CD40/CD40L in the CD4-dependent generation of CD8+ T cell immunity. J. Leukoc. Biol. 2000, 67, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Li, C.; Zhao, F.S.; Zhang, L.; Ng, T.B.; Jin, G.; Sha, O. Anti-tumor activity of toll-like receptor 7 agonists. Front. Pharmacol. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.M.; Morales, A.; Lamm, D.L. Bacillus Calmette-Gue´rin immunotherapy for genitourinary cancer. BJU Int. 2013, 112, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Tselikas, L.; de Baere, T.; Houot, R. Intratumoral immunotherapy: Using the tumor as the remedy. Ann. Oncol. 2017, 28, xii33–xii43. [Google Scholar] [CrossRef]
- Marabelle, A.; Kohrt, H.; Sagiv-Barfi, I.; Ajami, B.; Axtell, R.C.; Zhou, G.; Rajapaksa, R.; Green, M.R.; Torchia, J.; Brody, J.; et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J. Clin. Investig. 2013, 123, 2447–2463. [Google Scholar] [CrossRef] [PubMed]
- Houot, R.; Levy, R. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood 2009, 113, 3546–3552. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Grimm, E.; Heimberger, A.B. Innate immune functions of microglia isolated from human glioma patients. J. Transl. Med. 2006, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Glas, M.; Coch, C.; Trageser, D.; Daßler, J.; Simon, M.; Koch, P.; Mertens, J.; Quandel, T.; Gorris, R.; Reinartz, R.; et al. Targeting the cytosolic innate immune receptors RIG-I and MDA5 effectively counteracts cancer cell heterogeneity in glioblastoma. Stem. Cells 2013, 31, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Xun, Y.; Yang, H.; Kaminska, B.; You, H. Toll-like receptors and toll-like receptor-targeted immunotherapy against glioma. J. Hematol. Oncol. 2021, 14, 176. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma-are we there yet? Neuro. Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Abruzzese, M.P.; Bilotta, M.T.; Fionda, C.; Zingoni, A.; Soriani, A.; Vulpis, E.; Borrelli, C.; Zitti, B.; Petrucci, M.T.; Ricciardi, M.R.; et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of cMYC-IRF4-miR-125b interplay. J. Hematol. Oncol. 2016, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Knelson, E.H.; Jimenez-Macias, J.L.; Nowicki, M.O.; Han, S.; Panagioti, E.; Lizotte, P.H.; Adu-Berchie, K.; Stafford, A.; Dimitrakakis, N.; et al. STING activation promotes robust immune response and NK cell-mediated tumor regression in glioblastoma models. Proc. Natl. Acad. Sci. USA 2022, 119, e2111003119. [Google Scholar] [CrossRef]
- Boudreau, C.E.; Najem, H.; Ott, M.; Horbinski, C.; Fang, D.; DeRay, C.M.; Levine, J.M.; Curran, M.A.; Heimberger, A.B. Intratumoral Delivery of STING Agonist Results in Clinical Responses in Canine Glioblastoma. Clin. Cancer Res. 2021, 27, 5528–5535. [Google Scholar] [CrossRef]
- Hege, K.M.; Jooss, K.; Pardoll, D. GM-CSF gene-modifed cancer cell immunotherapies: Of mice and men. Int. Rev. Immunol. 2006, 25, 321–352. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Faisal, S.M.; Castro, M.G.; Lowenstein, P.R. Combined cytotoxic and immune-stimulatory gene therapy using Ad-TK and Ad-Flt3L: Translational developments from rodents to glioma patients. Mol. Ther. 2023, 31, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Correction to Combined cytotoxic and immune-stimulatory gene therapy for primary adult high-grade glioma: A phase 1, first-in-human trial. Lancet Oncol. 2023, 24, 1042–1052. [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.F.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type I IFN Contributes to NK Cell Homeostasis, Activation, and Antitumor Function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.F.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Rossi, E.A.; Rossi, D.L.; Cardillo, T.M.; Stein, R.; Goldenberg, D.M.; Chang, C.-H. Preclinical studies on targeted delivery of multiple IFNα2b to HLA-DR in diverse hematologic cancers. Blood 2011, 118, 1877–1884. [Google Scholar] [CrossRef]
- Cauwels, A.; Van Lint, S.; Paul, F.; Garcin, G.; De Koker, S.; Van Parys, A.; Wueest, T.; Gerlo, S.; Van der Heyden, J.; Bordat, Y.; et al. Delivering type i interferon to dendritic cells empowers tumor eradication and immune combination treatments. Cancer Res. 2018, 78, 463–474. [Google Scholar] [CrossRef]
- Vatner, R.E.; Janssen, E.M. STING, DCs and the link between innate and adaptive tumor immunity. Mol. Immunol. 2019, 110, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yang, Q.; Xu, P.; Deng, M.; Jiang, T.; Cai, L.; Li, J.; Sai, K.; Xi, S.; Ouyang, H.; et al. Adjuvant Temozolomide Chemotherapy with or Without Interferon Alfa among Patients with Newly Diagnosed High-grade Gliomas: A Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2253285. [Google Scholar] [CrossRef] [PubMed]
- Golán, I.; De La Fuente, L.R.; Costoya, J.A. NK cell-based glioblastoma immunotherapy. Cancers 2018, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Fehniger, T.A.; Ruggeri, L.; Velardi, A.; Caligiuri, M.A. Natural killer cell receptors: New biology and insights into the graft-versus-leukemia effect. Blood 2002, 100, 1935–1947. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Montemurro, F.; Aglietta, M. Trastuzumab: Mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann. Oncol. 2007, 18, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Albanell, J. Mechanism of action of anti-HER2 monoclonal antibodies. Ann. Oncol. 2001, 12, S35–S41. [Google Scholar] [CrossRef]
- Correale, P.; Botta, C.; Cusi, M.G.; Del Vecchio, M.; De Santi, M.; Savellini, G.G.; Bestoso, E.; Apollinari, S.; Mannucci, S.; Marra, M.; et al. Cetuximab ± chemotherapy enhances dendritic cell-mediated phagocytosis of colon cancer cells and ignites a highly efficient colon cancer antigen-specific cytotoxic T-cell response in vitro. Int. J. Cancer 2012, 130, 1577–1589. [Google Scholar] [CrossRef]
- Pozzi, C.; Cuomo, A.; Spadoni, I.; Magni, E.; Silvola, A.; Conte, A.; Sigismund, S.; Ravenda, P.S.; Bonaldi, T.; Zampino, M.G.; et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 2016, 22, 624–631. [Google Scholar] [CrossRef]
- Ren, Z.; Guo, J.; Liao, J.; Luan, Y.; Liu, Z.; Sun, Z.; Liu, X.; Liang, Y.; Peng, H.; Fu, Y.-X. CTLA-4 limits anti-CD20-mediated tumor regression. Clin. Cancer Res. 2017, 23, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Tian, L.; Chen, J.; Wang, J.; Ma, R.; Dong, W.; Li, A.; Zhang, J.; Chiocca, E.A.; Kaur, B.; et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat. Commun. 2021, 12, 5908. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα–CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-human, first-in-class phase i trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef]
- Lassman, A.B.; Van Den Bent, M.J.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR -amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro-Oncology 2019, 21, 106–114. [Google Scholar] [CrossRef]
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2020, 22, 684–693. [Google Scholar] [CrossRef]
- Mata-Molanes, J.J.; Rebollo-Liceaga, J.; Martínez-Navarro, E.M.; Manzano, R.G.; Brugarolas, A.; Juan, M.; Sureda, M. Relevance of Fc Gamma Receptor Polymorphisms in Cancer Therapy with Monoclonal Antibodies. Front. Oncol. 2022, 12, 926289. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Eng. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Z.; Fu, Y.X. Adapting conventional cancer treatment for immunotherapy. J. Mol. Med. 2016, 94, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic cancer vaccines. Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef]
- Patel, J.; Bozeman, E.N.; Selvaraj, P.; Bhargava, A.; Mishra, D.; Banerjee, S.; Mishra, P.K.; Kajihara, M.; Takakura, K.; Ohkusa, T.; et al. Taming dendritic cells with TIM-3: Another immunosuppressive strategy used by tumors. Immunotherapy 2012, 4, 1795–1798. [Google Scholar] [CrossRef]
- de Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103+ Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74.e6. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Lipson, E.J.; Tawbi, H.A.-H.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J. Clin. Oncol. 2021, 39, 9503. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse, I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Fares, J.; Davis, Z.B.; Rechberger, J.S.; Toll, S.A.; Schwartz, J.D.; Daniels, D.J.; Miller, J.S.; Khatua, S. Advances in NK cell therapy for brain tumors. NPJ Precis. Oncol. 2023, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.-T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef]
- Ohno, M.; Natsume, A.; Ichiro Iwami, K.; Iwamizu, H.; Noritake, K.; Ito, D.; Toi, Y.; Ito, M.; Motomura, K.; Yoshida, J.; et al. Retrovirally engineered T-cell-based immunotherapy targeting type III variant epidermal growth factor receptor, a glioma-associated antigen. Cancer Sci. 2010, 101, 2518–2524. [Google Scholar] [CrossRef]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. 2014, 20, 972–984. [Google Scholar] [CrossRef]
- Liu, X.; Song, J.; Zhang, H.; Liu, X.; Zuo, F.; Zhao, Y.; Zhao, Y.; Yin, X.; Guo, X.; Wu, X.; et al. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell 2023, 41, 272–287.e9. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1226720. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, e1226720. [Google Scholar] [CrossRef] [PubMed]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; André, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755.e15. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Z.; Xiang, G.L.; Zhu, X.H.; Xiu, L.; Sun, J.; Zhang, X. Advances in the mechanisms of action of cancer-targeting oncolytic viruses (review). Oncol. Lett. 2018, 15, 4053–4060. [Google Scholar] [CrossRef]
- Senior, M. Checkpoint inhibitors go viral. Nat. Biotechnol. 2019, 37, 12–17. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. OncoImmunology 2018, 7, e1442169. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef]
- de Graaf, J.F.; de Vor, L.; Fouchier, R.A.M.; van den Hoogen, B.G. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Gállego Pérez-Larraya, J.; Garcia-Moure, M.; Labiano, S.; Patiño-García, A.; Dobbs, J.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; Puigdelloses, M.; et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. N. Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. Innate Immune Response to Adenoviral Vectors Is Mediated by both Toll-Like Receptor-Dependent and -Independent Pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.; Stuart, J.; Stewart, R.M.R.; Berry, J.T.; Mainou, B.; Boehme, K.W. Current understanding of reovirus oncolysis mechanisms. Oncolytic. Virother. 2018, 7, 53–63. [Google Scholar] [CrossRef]
- Yount, J.S.; Moran, T.M.; López, C.B. Cytokine-Independent Upregulation of MDA5 in Viral Infection. J. Virol. 2007, 81, 7316–7319. [Google Scholar] [CrossRef] [PubMed]
- Hochrein, H.; Schlatter, B.; O’Keeffe, M.; Wagner, C.; Schmitz, F.; Schiemann, M.; Bauer, S.; Suter, M.; Wagner, H. Herpes simplex virus type-1 induces IFN-α production via Toll-like receptor 9-dependent and-independent pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 11416–11421. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic viral therapy and the immune system: A double-edged sword against cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef]
- Hines, J.B.; Kacew, A.J.; Sweis, R.F. The Development of STING Agonists and Emerging Results as a Cancer Immunotherapy. Curr. Oncol. Rep. 2023, 25, 189–199. [Google Scholar] [CrossRef]
- Ghaffari, A.; Peterson, N.; Khalaj, K.; Vitkin, N.; Robinson, A.; Francis, J.-A.; Koti, M. Sting agonist therapy in combination with pd-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br. J. Cancer 2018, 119, 440–449. [Google Scholar] [CrossRef]
- Moesta, A.K.; Cooke, K.; Piasecki, J.; Mitchell, P.; Rottman, J.B.; Fitzgerald, K.; Zhan, J.; Yang, B.; Le, T.; Belmontes, B.; et al. Local delivery of OncoVEXmGM-CSF generates systemic antitumor immune responses enhanced by cytotoxic T-lymphocyte–associated protein blockade. Clin. Cancer Res. 2017, 23, 6190–6202. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef]
- Rosewell Shaw, A.; Suzuki, M. Oncolytic Viruses Partner With T-Cell Therapy for Solid Tumor Treatment. Front. Immunol. 2018, 9, 2103. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Correction to: Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Watanabe, N.; McKenna, M.K.; Shaw, A.R.; Suzuki, M. Clinical CAR-T cell and oncolytic virotherapy for cancer treatment. Mol. Ther. 2021, 29, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.H.; Gul, K.; Arshad, A.; Riaz, N.; Waheed, U.; Rauf, A.; Aldakheel, F.; Alduraywish, S.; Rehman, M.U.; Abdullah, M.; et al. Microbiota in cancer development and treatment. J. Cancer Res. Clin. Oncol. 2019, 145, 49–63. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Kroemer, G.; Zitvogel, L. The intestinal microbiota determines the clinical efficacy of immune checkpoint blockers targeting PD-1/PD-L1. Oncoimmunology 2018, 7, e1434468. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Najafi, S.; Majidpoor, J.; Mortezaee, K. The impact of microbiota on PD-1/PD-L1 inhibitor therapy outcomes: A focus on solid tumors. Life Sci. 2022, 310, 121138. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Suez, J.; Elinav, E. The interplay between the innate immune system and the microbiota. Curr. Opin. Immunol. 2014, 26, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, X.; Zhou, X.; Zhao, J.; Yang, H.; Wang, S.; Morse, M.A.; Wu, J.; Yuan, Y.; Li, S.; et al. Blood microbiota diversity determines response of advanced colorectal cancer to chemotherapy combined with adoptive T cell immunotherapy. Oncoimmunology 2021, 10, 1976953. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillard, A.G.; Shin, D.H.; Hampton, L.A.; Lopez-Rivas, A.; Parthasarathy, A.; Fueyo, J.; Gomez-Manzano, C. Targeting Innate Immunity in Glioma Therapy. Int. J. Mol. Sci. 2024, 25, 947. https://doi.org/10.3390/ijms25020947
Gillard AG, Shin DH, Hampton LA, Lopez-Rivas A, Parthasarathy A, Fueyo J, Gomez-Manzano C. Targeting Innate Immunity in Glioma Therapy. International Journal of Molecular Sciences. 2024; 25(2):947. https://doi.org/10.3390/ijms25020947
Chicago/Turabian StyleGillard, Andrew G., Dong Ho Shin, Lethan A. Hampton, Andres Lopez-Rivas, Akhila Parthasarathy, Juan Fueyo, and Candelaria Gomez-Manzano. 2024. "Targeting Innate Immunity in Glioma Therapy" International Journal of Molecular Sciences 25, no. 2: 947. https://doi.org/10.3390/ijms25020947
APA StyleGillard, A. G., Shin, D. H., Hampton, L. A., Lopez-Rivas, A., Parthasarathy, A., Fueyo, J., & Gomez-Manzano, C. (2024). Targeting Innate Immunity in Glioma Therapy. International Journal of Molecular Sciences, 25(2), 947. https://doi.org/10.3390/ijms25020947