

New Insights into Endogenous Retrovirus-K Transcripts in Amyotrophic Lateral Sclerosis
 , , and
, , and
Abstract
:1. Introduction
2. Results
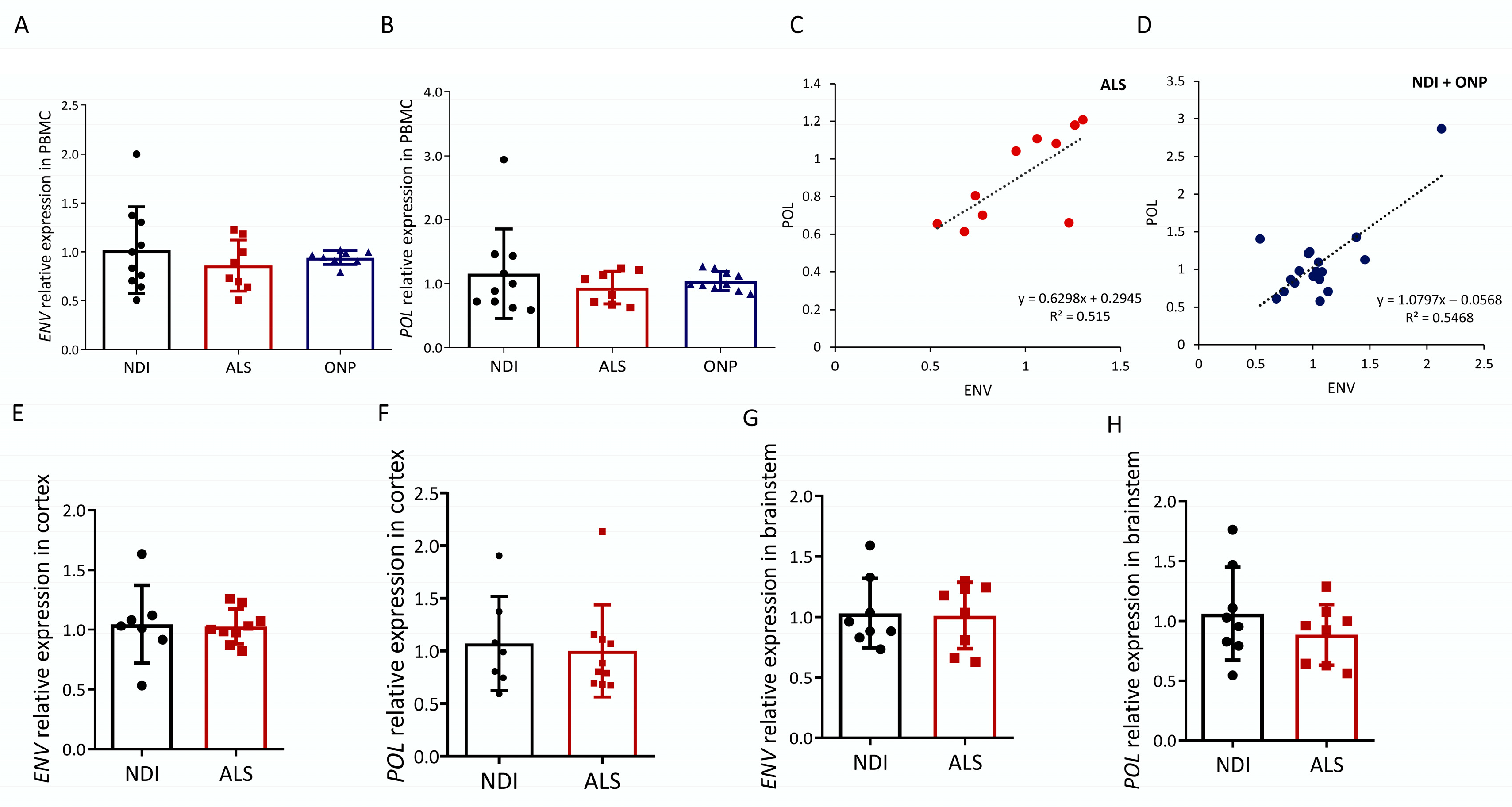
2.1. ALS-Associated HERVK Expression in PBMC
2.2. HERVK Expression in ALS Brain
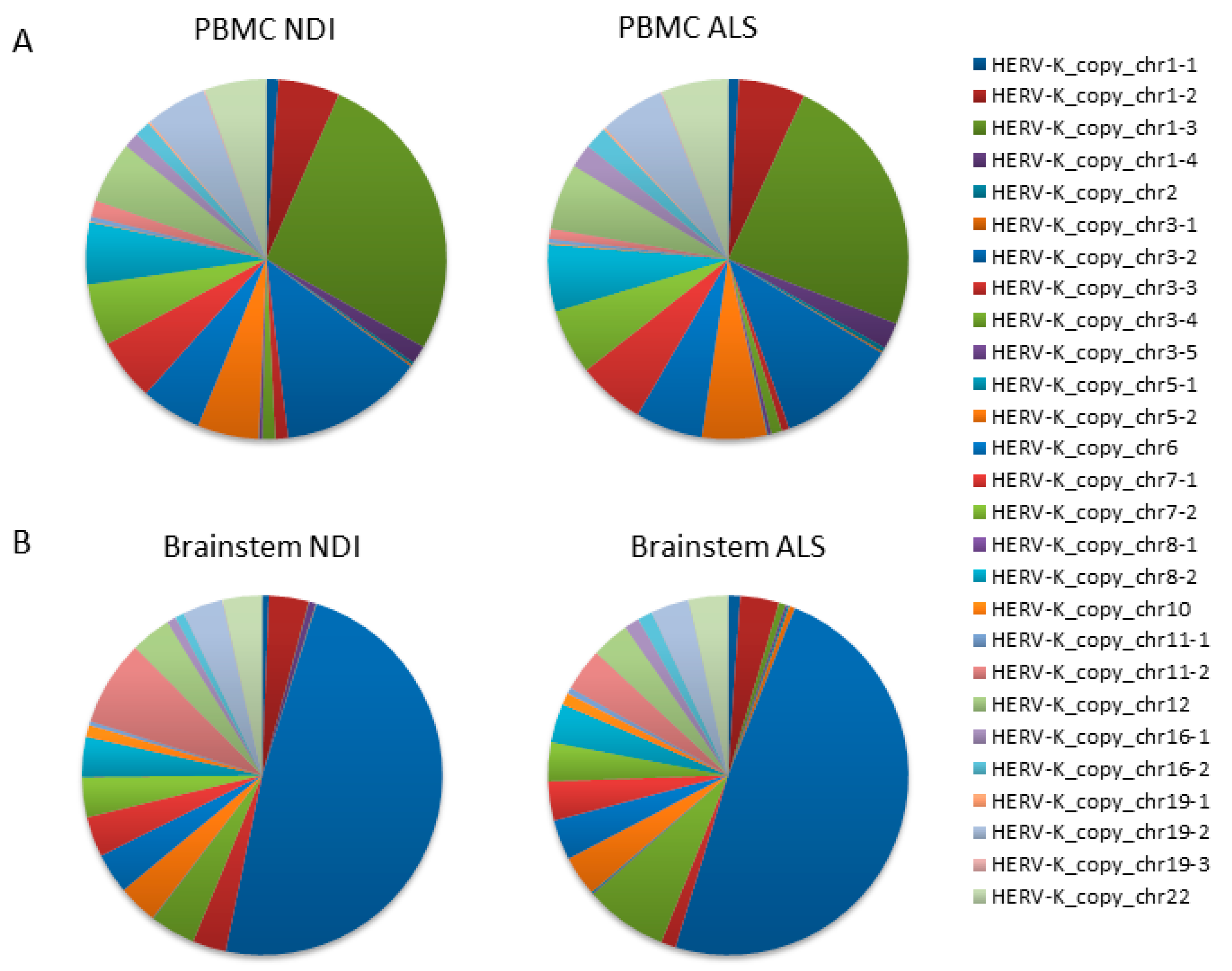
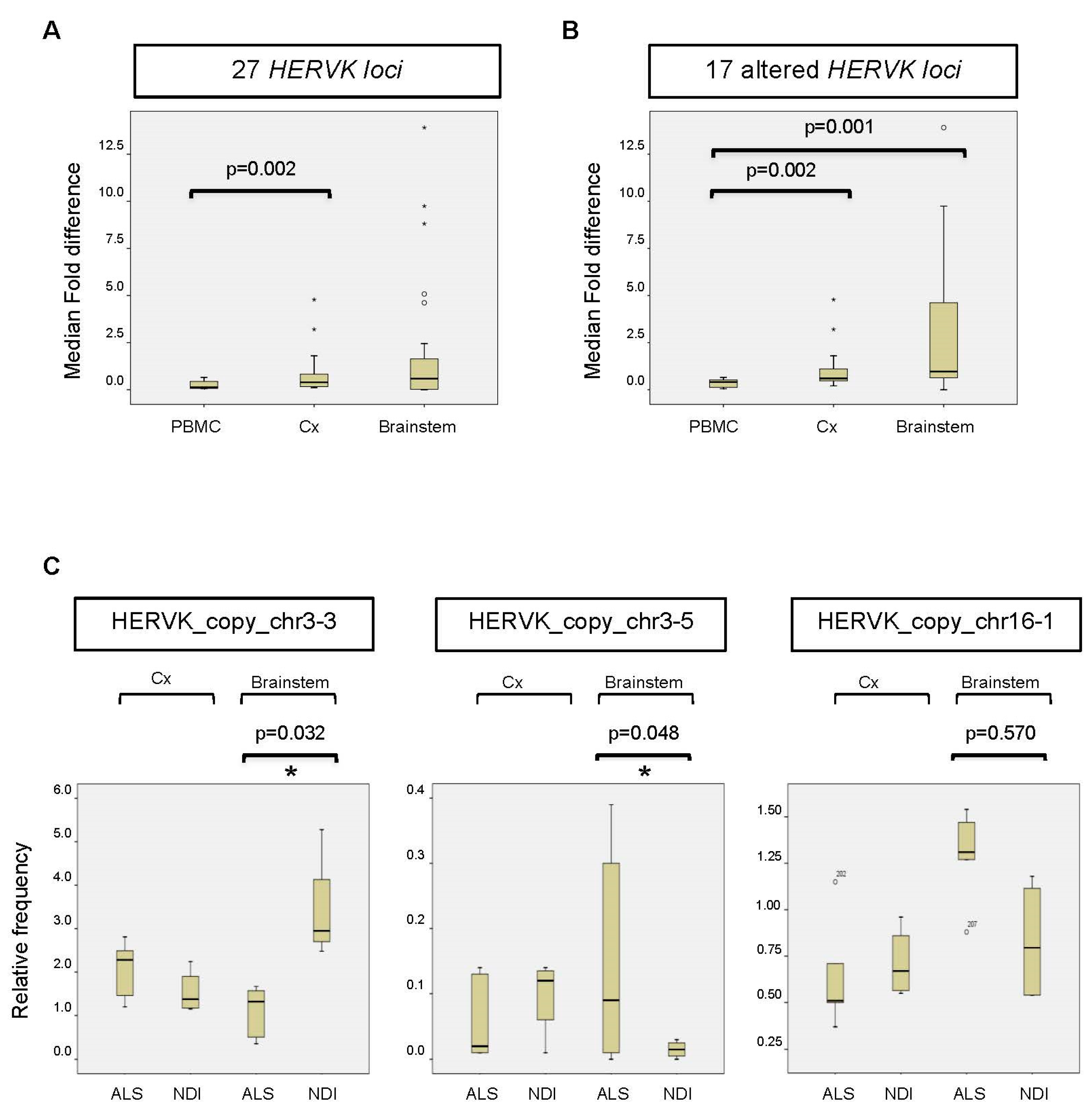
2.3. Analysis of Transcribed HERVK Loci
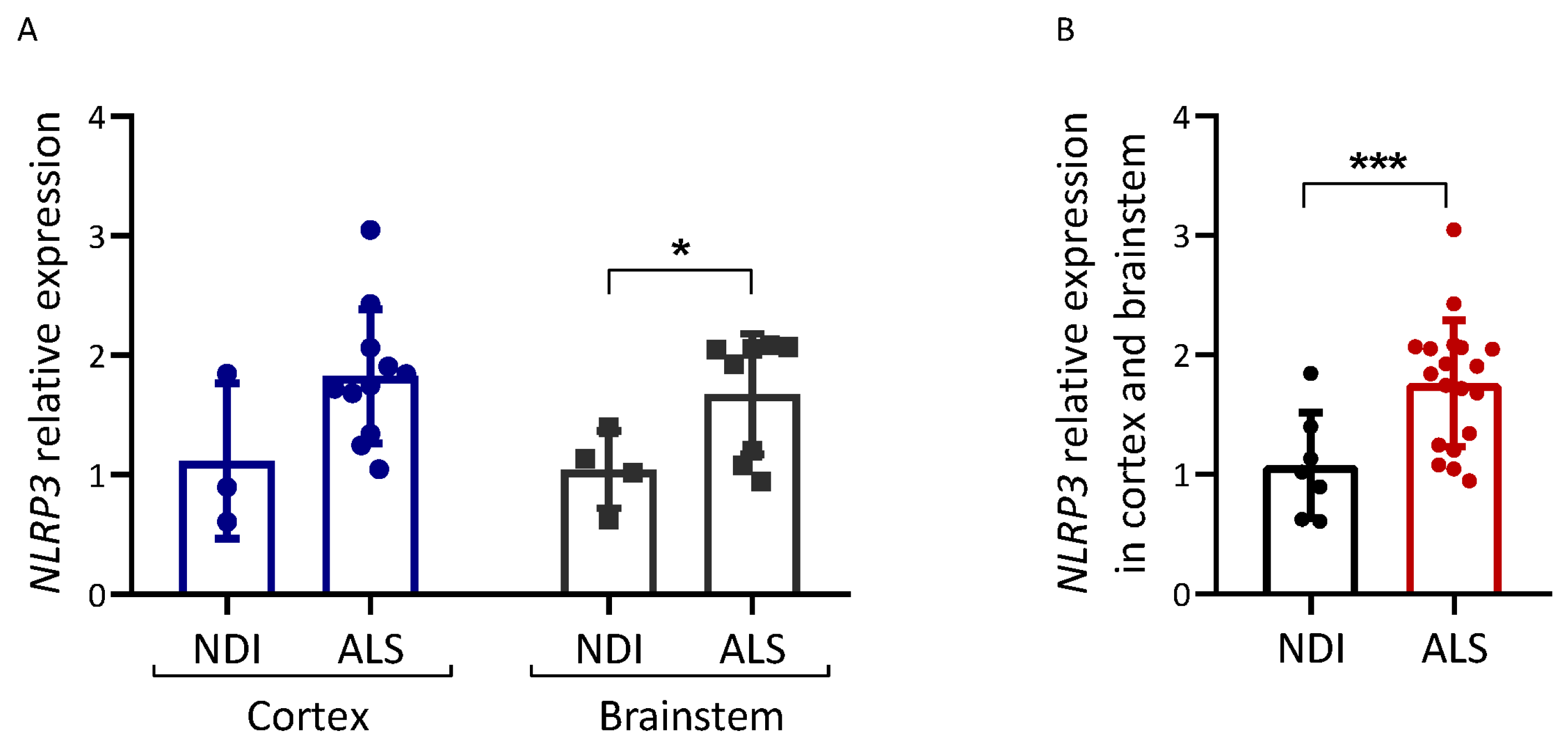
2.4. Inflammation Markers in ALS Brains
3. Discussion
4. Materials and Methods
4.1. Participants, Samples, and Approvals
4.2. Experimental Design
4.3. RNA Extraction, Preparation of cDNA, and qPCR
4.4. Next-Generation Sequencing (NGS)
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitchell, J.D.; Borasio, G.D. Amyotrophic Lateral Sclerosis. Lancet 2007, 369, 2031–2041. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical Genetics of Amyotrophic Lateral Sclerosis: What Do We Really Know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Puentes, F.; Malaspina, A.; van Noort, J.M.; Amor, S. Non-Neuronal Cells in ALS: Role of Glial, Immune Cells and Blood-CNS Barriers. Brain Pathol. 2016, 26, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Couratier, P.; Blasco, H.; Andres, C.R.; Beltran, S.; Meininger, V.; Vourc’h, P. Genetics of Amyotrophic Lateral Sclerosis. Rev. Neurol. 2017, 173, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.; O’Connell, J.D.; Yamazaki, T.; Gangopadhyay, J.; Gygi, S.P.; Reed, R. Interactome Analyses Revealed That the U1 SnRNP Machinery Overlaps Extensively with the RNAP II Machinery and Contains Multiple ALS/SMA-Causative Proteins. Sci. Rep. 2018, 8, 8755. [Google Scholar] [CrossRef]
- Gao, F.-B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated Molecular Pathways in Amyotrophic Lateral Sclerosis-Frontotemporal Dementia Spectrum Disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- McCombe, P.A.; Henderson, R.D. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Liu, C.-Y.; Che, C.-H.; Huang, H.-P. Toward Precision Medicine in Amyotrophic Lateral Sclerosis. Ann. Transl. Med. 2016, 4, 27. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M.; et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef]
- Hornung, V.; Latz, E. Critical Functions of Priming and Lysosomal Damage for NLRP3 Activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 Inflammasome Is Expressed by Astrocytes in the SOD1 Mouse Model of ALS and in Human Sporadic ALS Patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cao, C.; Qin, X.-Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased Peripheral Blood Inflammatory Cytokine Levels in Amyotrophic Lateral Sclerosis: A Meta-Analysis Study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Lieberman, J. Death by a Thousand Cuts: Granzyme Pathways of Programmed Cell Death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef] [PubMed]
- Iłżecka, J. Granzymes A and B Levels in Serum of Patients with Amyotrophic Lateral Sclerosis. Clin. Biochem. 2011, 44, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Schoorlemmer, J.; Pérez-Palacios, R.; Climent, M.; Guallar, D.; Muniesa, P. Regulation of Mouse Retroelement MuERV-L/MERVL Expression by REX1 and Epigenetic Control of Stem Cell Potency. Front. Oncol. 2014, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- de Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive Elements May Comprise over Two-Thirds of the Human Genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef]
- Dewannieux, M.; Heidmann, T. Endogenous Retroviruses: Acquisition, Amplification and Taming of Genome Invaders. Curr. Opin. Virol. 2013, 3, 646–656. [Google Scholar] [CrossRef]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, Characterization, and Comparative Genomic Distribution of the HERV-K (HML-2) Group of Human Endogenous Retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef]
- Kõks, S.; Kõks, G. The Role of Human Endogenous Retroviruses (HERVs) in the Pathologies of the Nervous System. In Molecular-Genetic and Statistical Techniques for Behavioral and Neural Research; Gerlai, R.T., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 519–533. ISBN 978-0-12-804078-2. [Google Scholar]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.-P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.H. Repetitive DNA in Disease. Science 2022, 376, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Phan, K.; He, Y.; Fu, Y.; Dzamko, N.; Bhatia, S.; Gold, J.; Rowe, D.; Ke, Y.D.; Ittner, L.M.; Hodges, J.R.; et al. Pathological Manifestation of Human Endogenous Retrovirus K in Frontotemporal Dementia. Commun. Med. 2021, 1, 60. [Google Scholar] [CrossRef]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human Endogenous Retrovirus-K Contributes to Motor Neuron Disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Steiner, J.P.; Bachani, M.; Malik, N.; DeMarino, C.; Li, W.; Sampson, K.; Lee, M.H.; Kowalak, J.; Bhaskar, M.; Doucet-O’Hare, T.; et al. Human Endogenous Retrovirus K Envelope in Spinal Fluid of Amyotrophic Lateral Sclerosis Is Toxic. Ann. Neurol. 2022, 92, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.J.; Al-Chalabi, A.; Ferrante, K.; Cudkowicz, M.E.; Brown, R.H.J.; Garson, J.A. Detection of Serum Reverse Transcriptase Activity in Patients with ALS and Unaffected Blood Relatives. Neurology 2005, 64, 454–458. [Google Scholar] [CrossRef]
- McCormick, A.L.; Brown, R.H.J.; Cudkowicz, M.E.; Al-Chalabi, A.; Garson, J.A. Quantification of Reverse Transcriptase in ALS and Elimination of a Novel Retroviral Candidate. Neurology 2008, 70, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Pandya, D.; Pasternack, N.; Garcia-Montojo, M.; Henderson, L.; Kozak, C.A.; Nath, A. Retroviral Elements in Pathophysiology and as Therapeutic Targets for Amyotrophic Lateral Sclerosis. Neurotherapeutics 2022, 19, 1085–1101. [Google Scholar] [CrossRef]
- Gold, J.; Rowe, D.B.; Kiernan, M.C.; Vucic, S.; Mathers, S.; van Eijk, R.P.A.; Nath, A.; Garcia Montojo, M.; Norato, G.; Santamaria, U.A.; et al. Safety and Tolerability of Triumeq in Amyotrophic Lateral Sclerosis: The Lighthouse Trial. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 595–604. [Google Scholar] [CrossRef]
- Garcia-Montojo, M.; Fathi, S.; Norato, G.; Smith, B.R.; Rowe, D.B.; Kiernan, M.C.; Vucic, S.; Mathers, S.; van Eijk, R.P.A.; Santamaria, U.; et al. Inhibition of HERV-K (HML-2) in Amyotrophic Lateral Sclerosis Patients on Antiretroviral Therapy. J. Neurol. Sci. 2021, 423, 117358. [Google Scholar] [CrossRef]
- Ishihara, T.; Koyama, A.; Hatano, Y.; Takeuchi, R.; Koike, Y.; Kato, T.; Tada, M.; Kakita, A.; Onodera, O. Endogenous Human Retrovirus-K Is Not Increased in the Affected Tissues of Japanese ALS Patients. Neurosci. Res. 2022, 178, 78–82. [Google Scholar] [CrossRef]
- Mayer, J.; Harz, C.; Sanchez, L.; Pereira, G.C.; Maldener, E.; Heras, S.R.; Ostrow, L.W.; Ravits, J.; Batra, R.; Meese, E.; et al. Transcriptional Profiling of HERV-K(HML-2) in Amyotrophic Lateral Sclerosis and Potential Implications for Expression of HML-2 Proteins. Mol. Neurodegener. 2018, 13, 39. [Google Scholar] [CrossRef]
- Garson, J.A.; Usher, L.; Al-Chalabi, A.; Huggett, J.; Day, E.F.; McCormick, A.L. Quantitative Analysis of Human Endogenous Retrovirus-K Transcripts in Postmortem Premotor Cortex Fails to Confirm Elevated Expression of HERV-K RNA in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2019, 7, 45. [Google Scholar] [CrossRef]
- Saresella, M.; Piancone, F.; Tortorella, P.; Marventano, I.; Gatti, A.; Caputo, D.; Lunetta, C.; Corbo, M.; Rovaris, M.; Clerici, M. T Helper-17 Activation Dominates the Immunologic Milieu of Both Amyotrophic Lateral Sclerosis and Progressive Multiple Sclerosis. Clin. Immunol. 2013, 148, 79–88. [Google Scholar] [CrossRef]
- Tamouza, R.; Meyer, U.; Lucas, A.; Richard, J.R.; Nkam, I.; Pinot, A.; Djonouma, N.; Boukouaci, W.; Charvet, B.; Pierquin, J.; et al. Patients with Psychosis Spectrum Disorders Hospitalized during the COVID-19 Pandemic Unravel Overlooked SARS-CoV-2 Past Infection Clustering with HERV-W ENV Expression and Chronic Inflammation. Transl. Psychiatry 2023, 13, 272. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Brunel, J.; Pierquin, J.; Iampietro, M.; Decimo, D.; Queruel, N.; Lucas, A.; Encabo-Berzosa, M.d.M.; Arenaz, I.; Marmolejo, T.P.; et al. SARS-CoV-2 Awakens Ancient Retroviral Genes and the Expression of Proinflammatory HERV-W Envelope Protein in COVID-19 Patients. iScience 2023, 26, 106604. [Google Scholar] [CrossRef]
- Garcia-Montojo, M.; Simula, E.R.; Fathi, S.; McMahan, C.; Ghosal, A.; Berry, J.D.; Cudkowicz, M.; Elkahloun, A.; Johnson, K.; Norato, G.; et al. Antibody Response to HML-2 May Be Protective in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2022, 92, 782–792. [Google Scholar] [CrossRef]
- Prudencio, M.; Gonzales, P.K.; Cook, C.N.; Gendron, T.F.; Daughrity, L.M.; Song, Y.; Ebbert, M.T.W.; van Blitterswijk, M.; Zhang, Y.-J.; Jansen-West, K.; et al. Repetitive Element Transcripts Are Elevated in the Brain of C9orf72 ALS/FTLD Patients. Hum. Mol. Genet. 2017, 26, 3421–3431. [Google Scholar] [CrossRef]
- Jones, A.R.; Iacoangeli, A.; Adey, B.N.; Bowles, H.; Shatunov, A.; Troakes, C.; Garson, J.A.; McCormick, A.L.; Al-Chalabi, A. A HML6 Endogenous Retrovirus on Chromosome 3 Is Upregulated in Amyotrophic Lateral Sclerosis Motor Cortex. Sci. Rep. 2021, 11, 14283. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. Locus Specific Reduction of L1 Expression in the Cortices of Individuals with Amyotrophic Lateral Sclerosis. Mol. Brain 2022, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.J.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Stetson, D.B. Endogenous Retroelements and Autoimmune Disease. Curr. Opin. Immunol. 2012, 24, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Cassel, S.L.; Sutterwala, F.S. Sterile Inflammatory Responses Mediated by the NLRP3 Inflammasome. Eur. J. Immunol. 2010, 40, 607–611. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Giacoppo, S.; Bramanti, P.; Mazzon, E. NLRP3 Inflammasome Activation in a Transgenic Amyotrophic Lateral Sclerosis Model. Inflammation 2018, 41, 93–103. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Lee, V.M.Y.; Trojanowski, J.Q. TDP-43 Functions and Pathogenic Mechanisms Implicated in TDP-43 Proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 Activates Microglia through NF-ΚB and NLRP3 Inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Leal-Lasarte, M.M.; Franco, J.M.; Labrador-Garrido, A.; Pozo, D.; Roodveldt, C. Extracellular TDP-43 Aggregates Target MAPK/MAK/MRK Overlapping Kinase (MOK) and Trigger Caspase-3/IL-18 Signaling in Microglia. FASEB J. 2017, 31, 2797–2816. [Google Scholar] [CrossRef]
- Rawat, P.; Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type-1 Single-Stranded RNA Activates the NLRP3 Inflammasome and Impairs Autophagic Clearance of Damaged Mitochondria in Human Microglia. Glia 2019, 67, 802–824. [Google Scholar] [CrossRef] [PubMed]
- Krestel, H.; Meier, J.C. RNA Editing and Retrotransposons in Neurology. Front. Mol. Neurosci. 2018, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.C.; Cibreiro, G.A.; Merino, P.T.; Roy, J.F.; Galiana, A.; Rufián, A.J.; Cano, J.M.; Martín, M.A.; Moreno, L.; Larrodé, P.; et al. Collagen XIX Alpha 1 Improves Prognosis in Amyotrophic Lateral Sclerosis. Aging Dis. 2019, 10, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Oros, D.; Strunk, M.; Breton, P.; Paules, C.; Benito, R.; Moreno, E.; Garcés, M.; Godino, J.; Schoorlemmer, J. Altered Gene Expression in Human Placenta after Suspected Preterm Labour. Placenta 2017, 55, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | fALS Patients (n = 10) | ONP Patients (n = 10) | NDIs (n = 10) |
|---|---|---|---|
| Gender (n) | 6 males 4 females | 6 males 4 females | 4 males 6 females |
| Age at illness onset (mean ± SD) | 60.04 ± 11.11 | ||
| Disease duration, months (mean ± SD) | 34.27 ± 20.73 | ||
| Age at sampling (mean ± SD) | 66.89 ± 11.61 | 52.37 ± 11.61 | 56.97 ± 10.55 |
| Patient Characteristics | sALS Patients (n = 10) | NDI (n = 9) |
| Brain anatomical region (n) | 10 frontal cortexes 8 brainstems | 6 frontal cortexes 8 brainstems |
| Gender (n) | 6 males 4 females | 6 males 3 females |
| Age at sampling (mean ± SD) | 58.64 ± 8.37 | 59.00 ± 8.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Martinez, L.; Macías-Redondo, S.; Strunk, M.; Guillén-Antonini, M.I.; Lunetta, C.; Tarlarini, C.; Penco, S.; Calvo, A.C.; Osta, R.; Schoorlemmer, J. New Insights into Endogenous Retrovirus-K Transcripts in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2024, 25, 1549. https://doi.org/10.3390/ijms25031549
Moreno-Martinez L, Macías-Redondo S, Strunk M, Guillén-Antonini MI, Lunetta C, Tarlarini C, Penco S, Calvo AC, Osta R, Schoorlemmer J. New Insights into Endogenous Retrovirus-K Transcripts in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2024; 25(3):1549. https://doi.org/10.3390/ijms25031549
Chicago/Turabian StyleMoreno-Martinez, Laura, Sofía Macías-Redondo, Mark Strunk, María Isabel Guillén-Antonini, Christian Lunetta, Claudia Tarlarini, Silvana Penco, Ana Cristina Calvo, Rosario Osta, and Jon Schoorlemmer. 2024. "New Insights into Endogenous Retrovirus-K Transcripts in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 25, no. 3: 1549. https://doi.org/10.3390/ijms25031549
APA StyleMoreno-Martinez, L., Macías-Redondo, S., Strunk, M., Guillén-Antonini, M. I., Lunetta, C., Tarlarini, C., Penco, S., Calvo, A. C., Osta, R., & Schoorlemmer, J. (2024). New Insights into Endogenous Retrovirus-K Transcripts in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 25(3), 1549. https://doi.org/10.3390/ijms25031549