
Neuroprotection via Carbon Monoxide Depends on the Circadian Regulation of CD36-Mediated Microglial Erythrophagocytosis in Hemorrhagic Stroke

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
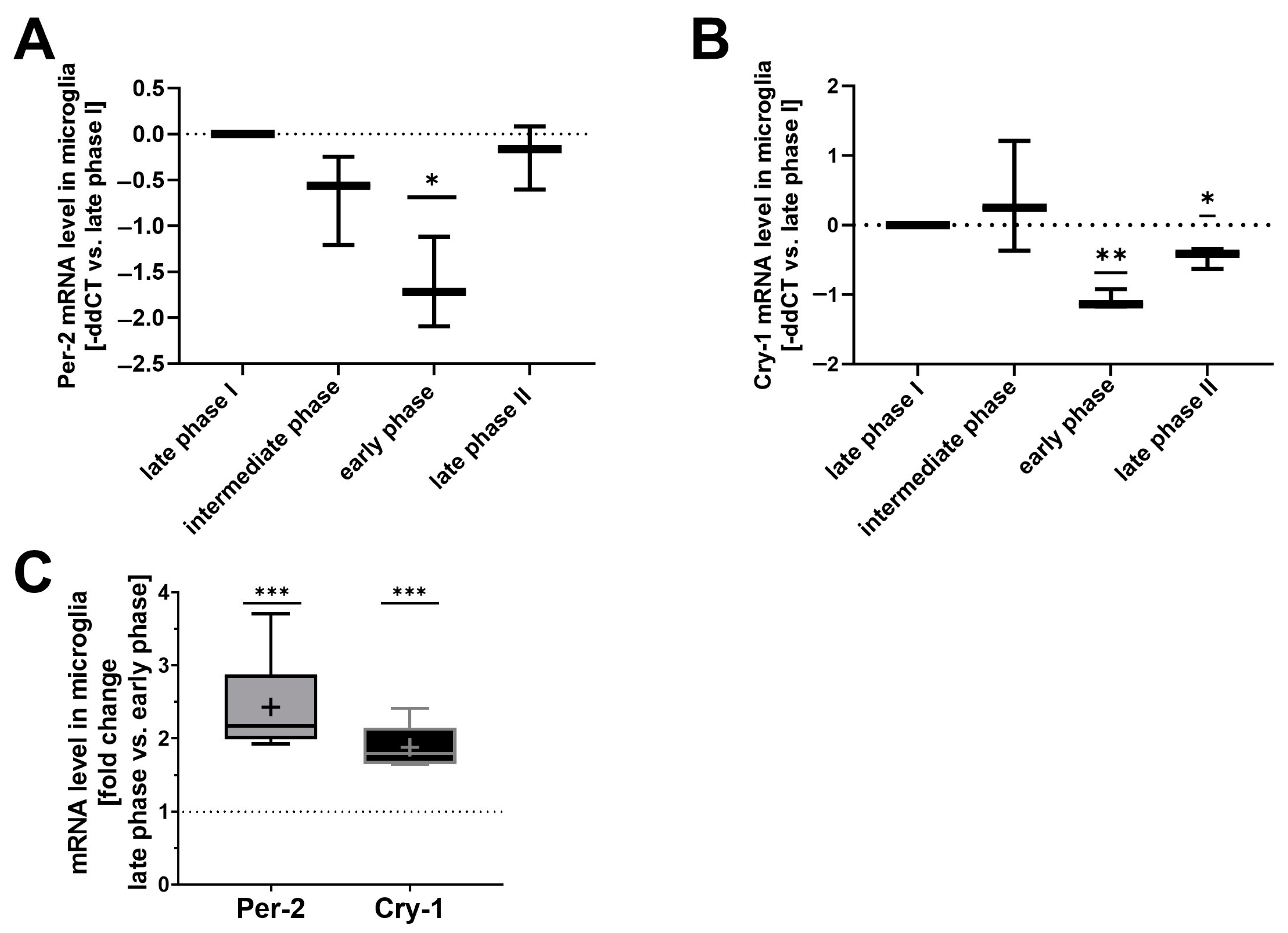
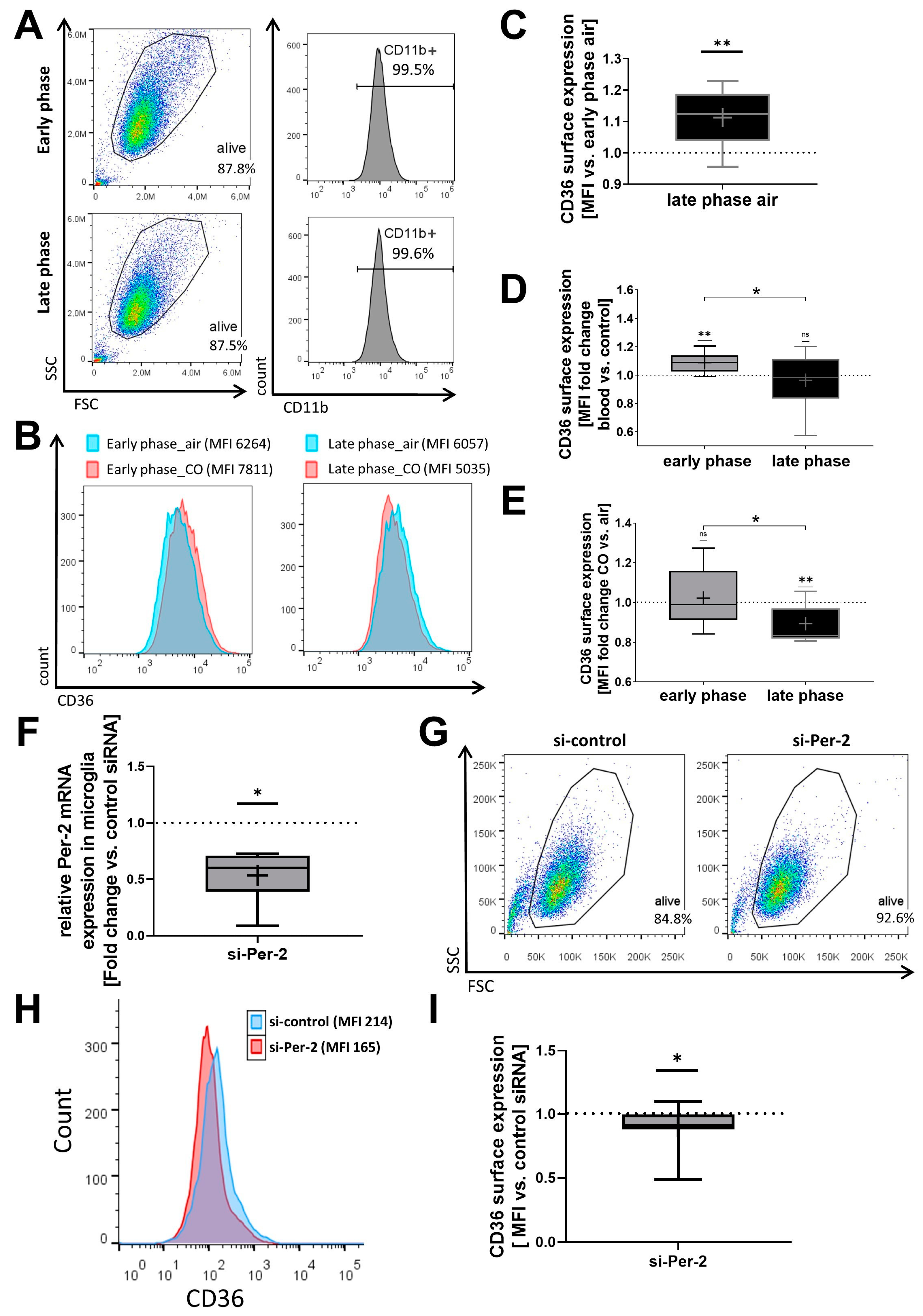
2.1. Circadian Gene Expression Profiles Determine CO-Mediated Induction of CD36 Surface Expression In Vitro
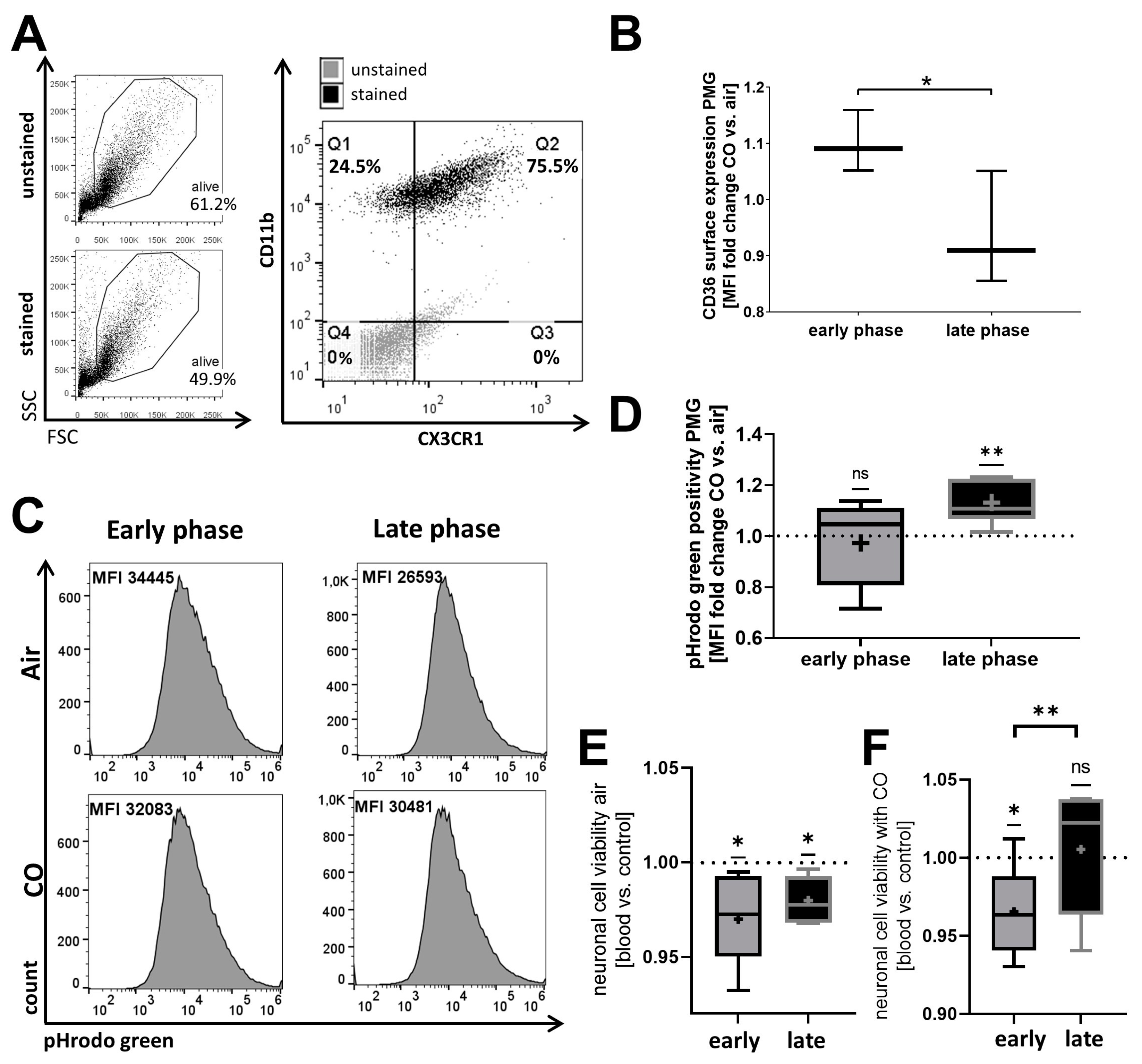
2.2. The Phagocytic Function of Primary Microglia (PMG) and Their Responsiveness to Exogenous CO Is Circadian-Dependent In Vitro
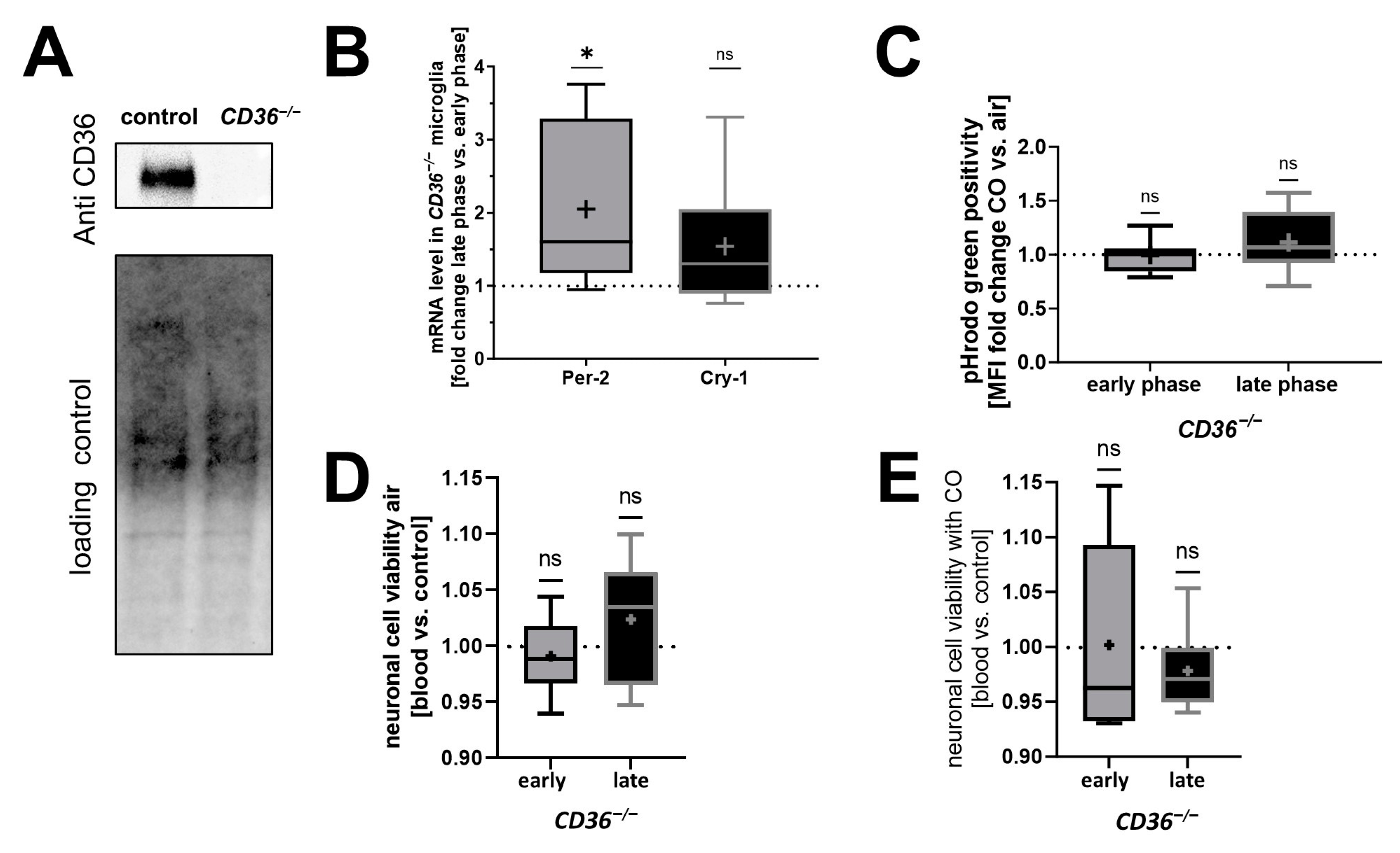
2.3. CD36−/− PMG Lack Circadian Rhythmicity and Responsiveness to CO Exposure In Vitro
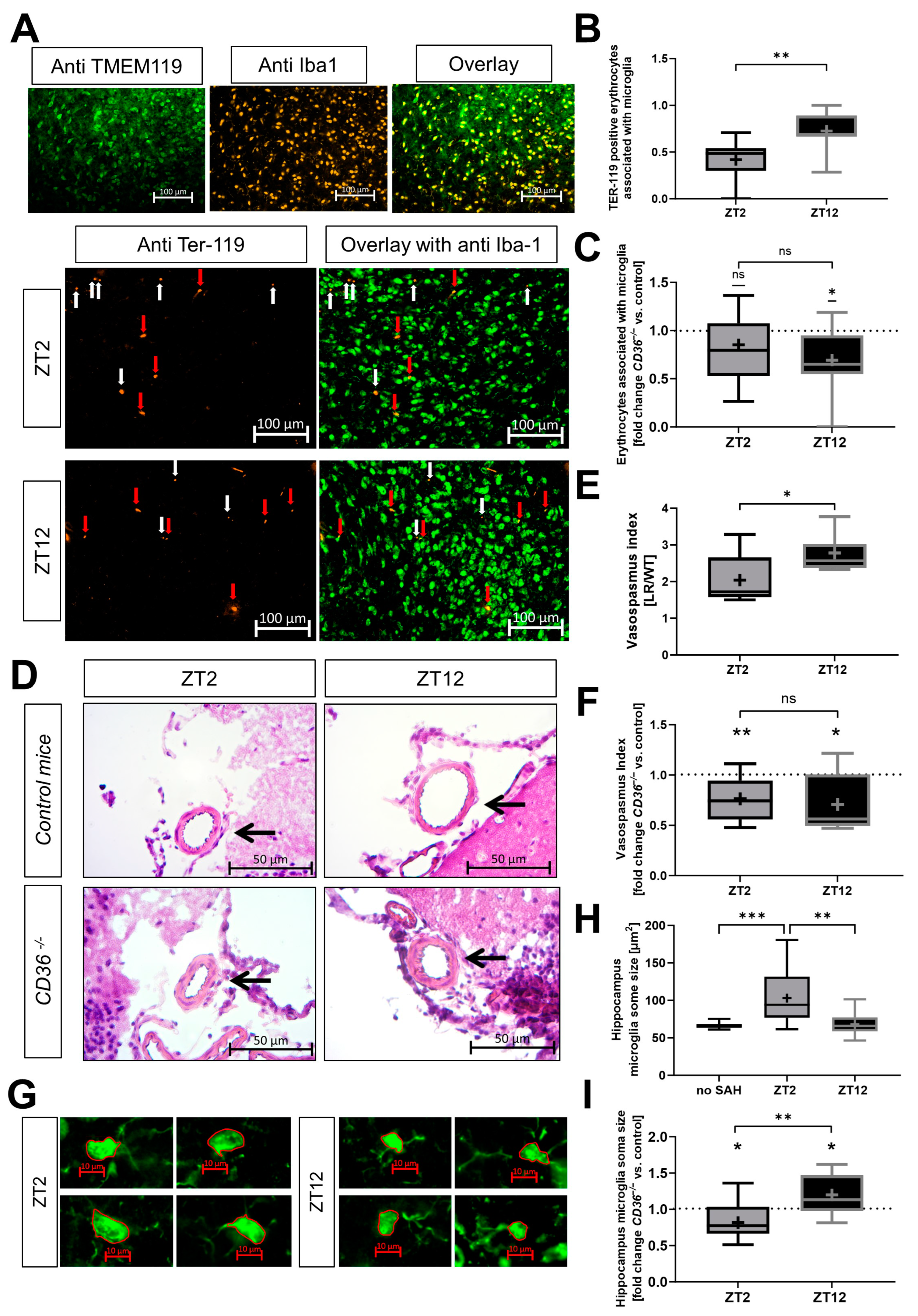
2.4. Erythrophagocytosis and Neuroinflammation Depend on the Circadian Timing of Injury and Expression of CD36 In Vivo
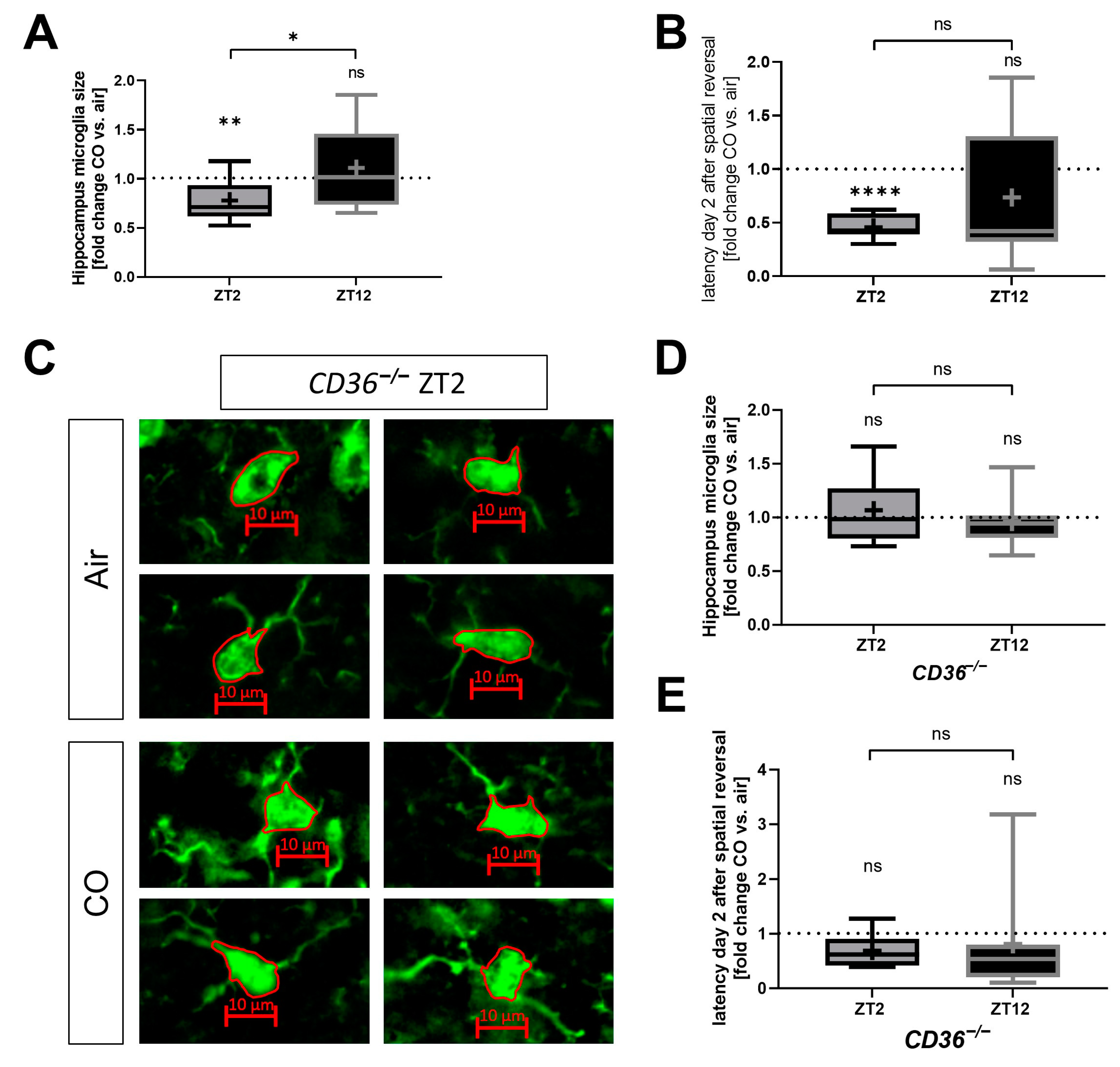
2.5. The Protective Effect of CO Depends on the Circadian Timing of Injury and Treatment
2.6. CO Does Not Protect CD36−/− Mice from Neuronal Injury Irrespective of Circadian Timing
3. Discussion
4. Materials and Methods
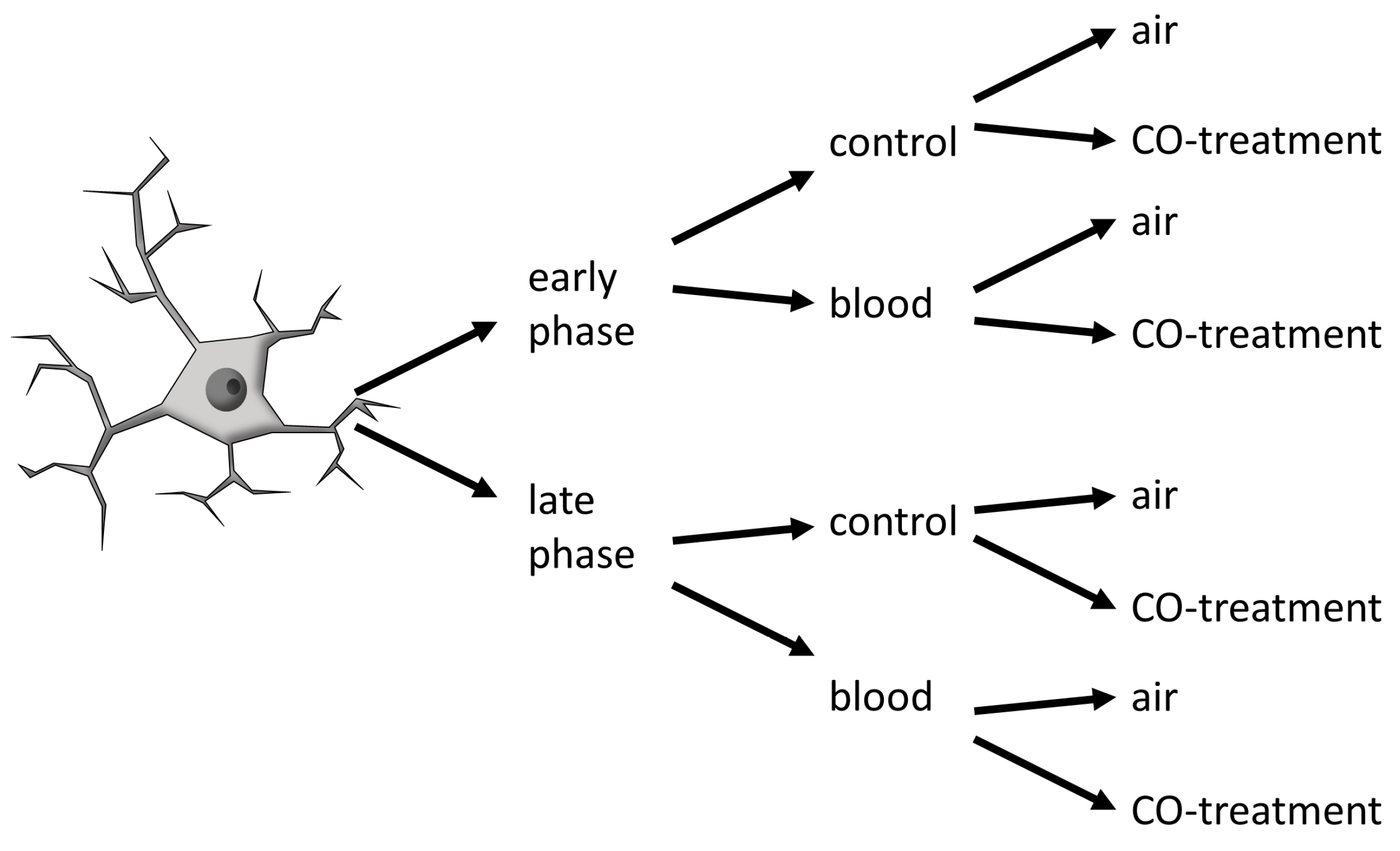
4.1. Animal Strains, Isolsated Microglia Cells and BV-2 Cellline in Culture
4.2. Circadian-Dependent Blood and CO Gas Treatment of Cells in Culture
4.3. Gene Expression Analyses from Cells and Tissue Samples
- Per-2
- 2.
- Cry-1
- 3.
- Rplp0
- 4.
- Per-2 (for siRNA experiments)
4.4. RNA Interference
4.5. Flow Cytometry
4.6. Western Blot Analysis
4.7. Co-Cultivation of PMG and HT22 for Viability Assay
4.8. Animal Care and Anesthesia
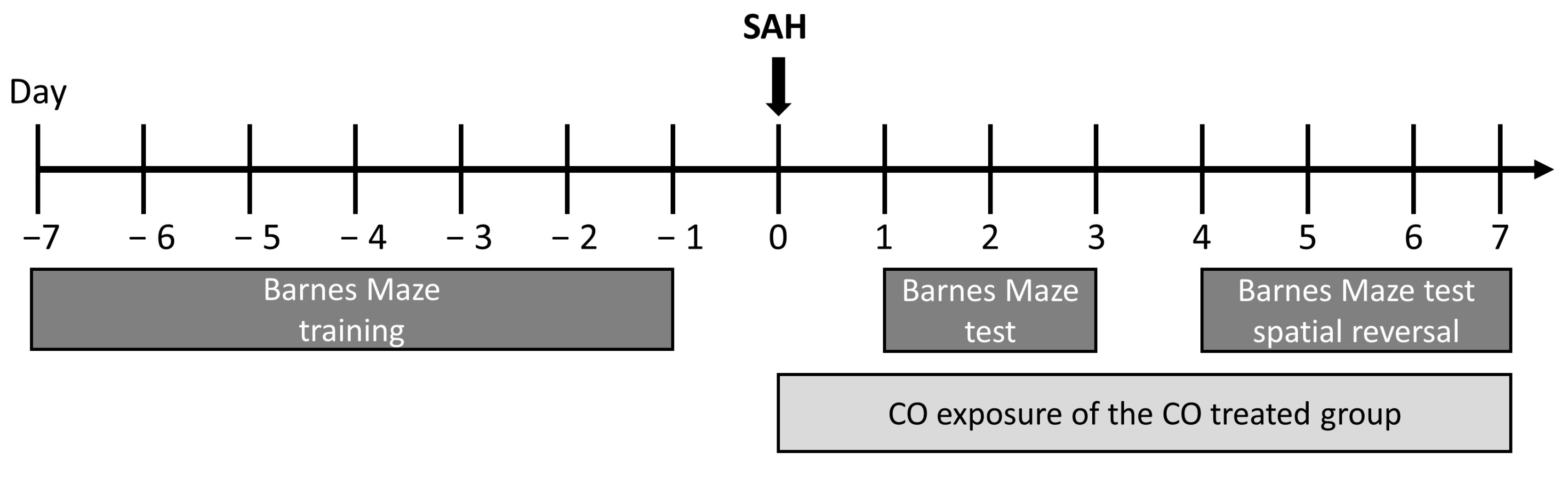
4.9. SAH Stroke Model
4.10. CO Gas Treatment of Mice
4.11. Hematoxylin/Eosin Staining and Evaluation of Cerebral Vasospasm
4.12. Immunohistochemistry
4.13. Barnes Maze Behavioral Studies
4.14. Statistics
4.15. Study Approval
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Temes, R.E.; Bleck, T.; Dugar, S.; Ouyang, B.; Mohammad, Y.; John, S.; Patel, P.; Lee, V.; Prabhakaran, S.; Quigg, M. Circadian variation in ictus of aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2012, 16, 219–223. [Google Scholar] [CrossRef]
- Chieregato, A.; Tagliaferri, F.; Cocciolo, F.; Boari, B.; Gallerani, M.; Manfredini, R. Can circadian rhythms influence onset and outcome of nontraumatic subarachnoid hemorrhage? Am. J. Emerg. Med. 2007, 25, 728–730. [Google Scholar] [CrossRef]
- Manfredini, R.; Boari, B.; Smolensky, M.H.; Salmi, R.; la Cecilia, O.; Maria Malagoni, A.; Haus, E.; Manfredini, F. Circadian variation in stroke onset: Identical temporal pattern in ischemic and hemorrhagic events. Chronobiol. Int. 2005, 22, 417–453. [Google Scholar] [CrossRef]
- Karatsoreos, I.N.; Bhagat, S.; Bloss, E.B.; Morrison, J.H.; McEwen, B.S. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, I.; Leinweber, B.; Brandenburger, M.; Oster, H. Circadian clock network desynchrony promotes weight gain and alters glucose homeostasis in mice. Mol. Metab. 2019, 30, 140–151. [Google Scholar] [CrossRef]
- Shearman, L.P.; Sriram, S.; Weaver, D.R.; Maywood, E.S.; Chaves, I.; Zheng, B.; Kume, K.; Lee, C.C.; van der Horst, G.T.; Hastings, M.H.; et al. Interacting molecular loops in the mammalian circadian clock. Science 2000, 288, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Hartmann, K.; Bonney, S.; Reithel, S.; Mittelbronn, M.; Walker, L.A.; Lowes, B.D.; Han, J.; Borchers, C.H.; Buttrick, P.M.; et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 2012, 18, 774–782. [Google Scholar] [CrossRef]
- Chen, P.; Li, C.; Pang, W.; Zhao, Y.; Dong, W.; Wang, S.; Zhang, J. The protective role of Per2 against carbon tetrachloride-induced hepatotoxicity. Am. J. Pathol. 2009, 174, 63–70. [Google Scholar] [CrossRef]
- Wiebking, N.; Maronde, E.; Rami, A. Increased neuronal injury in clock gene Per-1 deficient-mice after cerebral ischemia. Curr. Neurovasc. Res. 2013, 10, 112–125. [Google Scholar] [CrossRef]
- Schallner, N.; Lieberum, J.L.; Gallo, D.; LeBlanc, R.H., 3rd; Fuller, P.M.; Hanafy, K.A.; Otterbein, L.E. Carbon Monoxide Preserves Circadian Rhythm to Reduce the Severity of Subarachnoid Hemorrhage in Mice. Stroke A J. Cereb. Circ. 2017, 48, 2565–2573. [Google Scholar] [CrossRef]
- Schallner, N.; Pandit, R.; LeBlanc, R., 3rd; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.; Selzner, L.; Weber, J.; Schallner, N. Carbon monoxide controls microglial erythrophagocytosis by regulating CD36 surface expression to reduce the severity of hemorrhagic injury. Glia 2020, 68, 2427–2445. [Google Scholar] [CrossRef]
- McGilvray, I.D.; Serghides, L.; Kapus, A.; Rotstein, O.D.; Kain, K.C. Nonopsonic monocyte/macrophage phagocytosis of Plasmodium falciparum-parasitized erythrocytes: A role for CD36 in malarial clearance. Blood 2000, 96, 3231–3240. [Google Scholar] [CrossRef]
- Fang, H.; Chen, J.; Lin, S.; Wang, P.; Wang, Y.; Xiong, X.; Yang, Q. CD36-mediated hematoma absorption following intracerebral hemorrhage: Negative regulation by TLR4 signaling. J. Immunol. 2014, 192, 5984–5992. [Google Scholar] [CrossRef]
- Flores, J.J.; Klebe, D.; Rolland, W.B.; Lekic, T.; Krafft, P.R.; Zhang, J.H. PPARgamma-induced upregulation of CD36 enhances hematoma resolution and attenuates long-term neurological deficits after germinal matrix hemorrhage in neonatal rats. Neurobiol. Dis. 2016, 87, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Wang, L.; Hang, H.; Liu, C.; Wu, G. Rosiglitazone pretreatment influences thrombin-induced phagocytosis by rat microglia via activating PPARgamma and CD36. Neurosci. Lett. 2017, 651, 159–164. [Google Scholar] [CrossRef]
- Dioum, E.M.; Rutter, J.; Tuckerman, J.R.; Gonzalez, G.; Gilles-Gonzalez, M.A.; McKnight, S.L. NPAS2: A gas-responsive transcription factor. Science 2002, 298, 2385–2387. [Google Scholar] [CrossRef] [PubMed]
- Lukat-Rodgers, G.S.; Correia, C.; Botuyan, M.V.; Mer, G.; Rodgers, K.R. Heme-based sensing by the mammalian circadian protein CLOCK. Inorg. Chem. 2010, 49, 6349–6365. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Pozdeyev, N.; Haque, R.; Visser, A.; Ivanova, T.N.; Iuvone, P.M. Circadian clockwork machinery in neural retina: Evidence for the presence of functional clock components in photoreceptor-enriched chick retinal cell cultures. Mol. Vis. 2006, 12, 215–223. [Google Scholar]
- Soares, N.L.; Paiva, I.; Bravo, J.; Queiroga, C.S.F.; Melo, B.F.; Conde, S.V.; Romao, C.C.; Summavielle, T.; Vieira, H.L.A. Carbon Monoxide Modulation of Microglia-Neuron Communication: Anti-Neuroinflammatory and Neurotrophic Role. Mol. Neurobiol. 2022, 59, 872–889. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Ahmad, A.S.; Dore, S. Carbon monoxide attenuates vasospasm and improves neurobehavioral function after subarachnoid hemorrhage. Arch Biochem. Biophys 2019, 676, 108117. [Google Scholar] [CrossRef] [PubMed]
- Czeisler, C.A.; Duffy, J.F.; Shanahan, T.L.; Brown, E.N.; Mitchell, J.F.; Rimmer, D.W.; Ronda, J.M.; Silva, E.J.; Allan, J.S.; Emens, J.S.; et al. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science 1999, 284, 2177–2181. [Google Scholar] [CrossRef] [PubMed]
- Frase, S.; Kaiser, S.; Steimer, M.; Selzner, L.; Foit, N.A.; Niesen, W.D.; Schallner, N. Patients with Subarachnoid Hemorrhage Exhibit Disturbed Expression Patterns of the Circadian Rhythm Gene Period-2. Life 2021, 11, 124. [Google Scholar] [CrossRef]
- Suzuki, H.; Kawakita, F.; Asada, R. Neuroelectric Mechanisms of Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2022, 23, 3102. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, M.; Liu, Z.; Wang, Y.; Xiong, K. Molecular mechanisms of neuronal death in brain injury after subarachnoid hemorrhage. Front. Cell. Neurosci. 2022, 16, 1025708. [Google Scholar] [CrossRef]
- Solar, P.; Zamani, A.; Lakatosova, K.; Joukal, M. The blood-brain barrier and the neurovascular unit in subarachnoid hemorrhage: Molecular events and potential treatments. Fluids Barriers CNS 2022, 19, 29. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Li, Y.; Guo, Y.; Liu, B.; Tian, Y.; Wu, P.; Shi, H. Metformin attenuates early brain injury after subarachnoid hemorrhage in rats via AMPK-dependent mitophagy. Exp. Neurol. 2022, 353, 114055. [Google Scholar] [CrossRef]
- Li, R.; Zhao, M.; Yao, D.; Zhou, X.; Lenahan, C.; Wang, L.; Ou, Y.; He, Y. The role of the astrocyte in subarachnoid hemorrhage and its therapeutic implications. Front. Immunol. 2022, 13, 1008795. [Google Scholar] [CrossRef]
- Matz, P.; Turner, C.; Weinstein, P.R.; Massa, S.M.; Panter, S.S.; Sharp, F.R. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996, 713, 211–222. [Google Scholar] [CrossRef]
- Sutherland, B.A.; Rahman, R.M.; Clarkson, A.N.; Shaw, O.M.; Nair, S.M.; Appleton, I. Cerebral heme oxygenase 1 and 2 spatial distribution is modulated following injury from hypoxia-ischemia and middle cerebral artery occlusion in rats. Neurosci. Res. 2009, 65, 326–334. [Google Scholar] [CrossRef]
- Takeda, A.; Kimpara, T.; Onodera, H.; Itoyama, Y.; Shibahara, S.; Kogure, K. Regional difference in induction of heme oxygenase-1 protein following rat transient forebrain ischemia. Neurosci. Lett. 1996, 205, 169–172. [Google Scholar] [CrossRef]
- Turner, C.P.; Panter, S.S.; Sharp, F.R. Anti-oxidants prevent focal rat brain injury as assessed by induction of heat shock proteins (HSP70, HO-1/HSP32, HSP47) following subarachnoid injections of lysed blood. Brain Res. Mol. Brain Res. 1999, 65, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Tsunoda, H.; Zang, L.; Hirano, M.; Oka, T.; Tanaka, T. Synergistic induction of heme oxygenase-1 by nicaraven after subarachnoid hemorrhage to prevent delayed cerebral vasospasm. Eur. J. Pharmacol. 2009, 620, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, S.; Zhang, M.; Weng, Z.; Li, P.; Gan, Y.; Zhang, L.; Cao, G.; Gao, Y.; Leak, R.K.; et al. Pharmacological induction of heme oxygenase-1 by a triterpenoid protects neurons against ischemic injury. Stroke 2012, 43, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S.; Zhuang, H.; Biswal, S.; Christen, Y.; Dore, S. Ginkgo biloba extract neuroprotective action is dependent on heme oxygenase 1 in ischemic reperfusion brain injury. Stroke 2008, 39, 3389–3396. [Google Scholar] [CrossRef]
- Wang, H.Q.; Xu, Y.X.; Zhu, C.Q. Upregulation of heme oxygenase-1 by acteoside through ERK and PI3 K/Akt pathway confer neuroprotection against beta-amyloid-induced neurotoxicity. Neurotox. Res. 2012, 21, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Fang, Q.; Zhang, J.; Zhou, D.; Wang, Z. Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J. Neurosci. Res. 2011, 89, 515–523. [Google Scholar] [CrossRef]
- Foresti, R.; Bains, S.K.; Pitchumony, T.S.; de Castro Bras, L.E.; Drago, F.; Dubois-Rande, J.L.; Bucolo, C.; Motterlini, R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol. Res. 2013, 76, 132–148. [Google Scholar] [CrossRef]
- Li, F.; Faustino, J.; Woo, M.S.; Derugin, N.; Vexler, Z.S. Lack of the scavenger receptor CD36 alters microglial phenotypes after neonatal stroke. J. Neurochem. 2015, 135, 445–452. [Google Scholar] [CrossRef]
- Li, X.; Melief, E.; Postupna, N.; Montine, K.S.; Keene, C.D.; Montine, T.J. Prostaglandin E2 Receptor Subtype 2 Regulation of Scavenger Receptor CD36 Modulates Microglial Abeta42 Phagocytosis. Am. J. Pathol. 2015, 185, 230–239. [Google Scholar] [CrossRef]
- Yamanaka, M.; Ishikawa, T.; Griep, A.; Axt, D.; Kummer, M.P.; Heneka, M.T. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 2012, 32, 17321–17331. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Warner, M.L.; Bratton, D.L.; Henson, P.M. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3). J. Immunol. 1998, 161, 6250–6257. [Google Scholar] [CrossRef] [PubMed]
- Hajri, T.; Han, X.X.; Bonen, A.; Abumrad, N.A. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Investig. 2002, 109, 1381–1389. [Google Scholar] [CrossRef]
- Cifarelli, V.; Appak-Baskoy, S.; Peche, V.S.; Kluzak, A.; Shew, T.; Narendran, R.; Pietka, K.M.; Cella, M.; Walls, C.W.; Czepielewski, R.; et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat. Commun. 2021, 12, 3350. [Google Scholar] [CrossRef]
- Mistry, J.J.; Hellmich, C.; Moore, J.A.; Jibril, A.; Macaulay, I.; Moreno-Gonzalez, M.; Di Palm, F.; Beraza, N.; Bowles, K.M.; Rushworth, S.A. Free fatty-acid transport via CD36 drives beta-oxidation-mediated hematopoietic stem cell response to infection. Nat. Commun. 2021, 12, 7130. [Google Scholar] [CrossRef]
- Van Dycke, K.C.; Pennings, J.L.; van Oostrom, C.T.; van Kerkhof, L.W.; van Steeg, H.; van der Horst, G.T.; Rodenburg, W. Biomarkers for circadian rhythm disruption independent of time of day. PLoS ONE 2015, 10, e0127075. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Queiroz, J.; Hussain, M.M. Nonalcoholic fatty liver disease in CLOCK mutant mice. J. Clin. Investg. 2020, 130, 4282–4300. [Google Scholar] [CrossRef] [PubMed]
- Prabhat, A.; Malik, I.; Jha, N.A.; Bhardwaj, S.K.; Kumar, V. Developmental effects of constant light on circadian behaviour and gene expressions in zebra finches: Insights into mechanisms of metabolic adaptation to aperiodic environment in diurnal animals. J. Photochem. Photobiol. B 2020, 211, 111995. [Google Scholar] [CrossRef]
- Xu, W.; Xu, R.; Li, X.; Zhang, H.; Wang, X.; Zhu, J. Downregulating hypoxia-inducible factor-1alpha expression with perfluorooctyl-bromide nanoparticles reduces early brain injury following experimental subarachnoid hemorrhage in rats. Am. J. Transl. Res. 2016, 8, 2114–2126. [Google Scholar]
- Jin, J.; Duan, J.; Du, L.; Xing, W.; Peng, X.; Zhao, Q. Inflammation and immune cell abnormalities in intracranial aneurysm subarachnoid hemorrhage (SAH): Relevant signaling pathways and therapeutic strategies. Front. Immunol. 2022, 13, 1027756. [Google Scholar] [CrossRef]
- Peliciari-Garcia, R.A.; Bargi-Souza, P.; Young, M.E.; Nunes, M.T. Repercussions of hypo and hyperthyroidism on the heart circadian clock. Chronobiol. Int. 2018, 35, 147–159. [Google Scholar] [CrossRef]
- Prolo, L.M.; Takahashi, J.S.; Herzog, E.D. Circadian rhythm generation and entrainment in astrocytes. J. Neurosci. 2005, 25, 404–408. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Stupfel, M.; Bouley, G. Physiological and biochemical effects on rats and mice exposed to small concentrations of carbon monoxide for long periods. Ann. N. Y. Acad. Sci. 1970, 174, 342–368. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G.; Lam, F.; Hagen, T.; Moncada, S. Inhibition of cellular respiration by endogenously produced carbon monoxide. J. Cell Sci. 2006, 119 Pt 11, 2291–2298. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, S.; Henrich, L.; Kiessling, I.; Loy, B.; Schallner, N. Neuroprotection via Carbon Monoxide Depends on the Circadian Regulation of CD36-Mediated Microglial Erythrophagocytosis in Hemorrhagic Stroke. Int. J. Mol. Sci. 2024, 25, 1680. https://doi.org/10.3390/ijms25031680
Kaiser S, Henrich L, Kiessling I, Loy B, Schallner N. Neuroprotection via Carbon Monoxide Depends on the Circadian Regulation of CD36-Mediated Microglial Erythrophagocytosis in Hemorrhagic Stroke. International Journal of Molecular Sciences. 2024; 25(3):1680. https://doi.org/10.3390/ijms25031680
Chicago/Turabian StyleKaiser, Sandra, Luise Henrich, Iva Kiessling, Benedikt Loy, and Nils Schallner. 2024. "Neuroprotection via Carbon Monoxide Depends on the Circadian Regulation of CD36-Mediated Microglial Erythrophagocytosis in Hemorrhagic Stroke" International Journal of Molecular Sciences 25, no. 3: 1680. https://doi.org/10.3390/ijms25031680
APA StyleKaiser, S., Henrich, L., Kiessling, I., Loy, B., & Schallner, N. (2024). Neuroprotection via Carbon Monoxide Depends on the Circadian Regulation of CD36-Mediated Microglial Erythrophagocytosis in Hemorrhagic Stroke. International Journal of Molecular Sciences, 25(3), 1680. https://doi.org/10.3390/ijms25031680