
The African Swine Fever Virus Virulence Determinant DP96R Suppresses Type I IFN Production Targeting IRF3

 ,
, 
 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
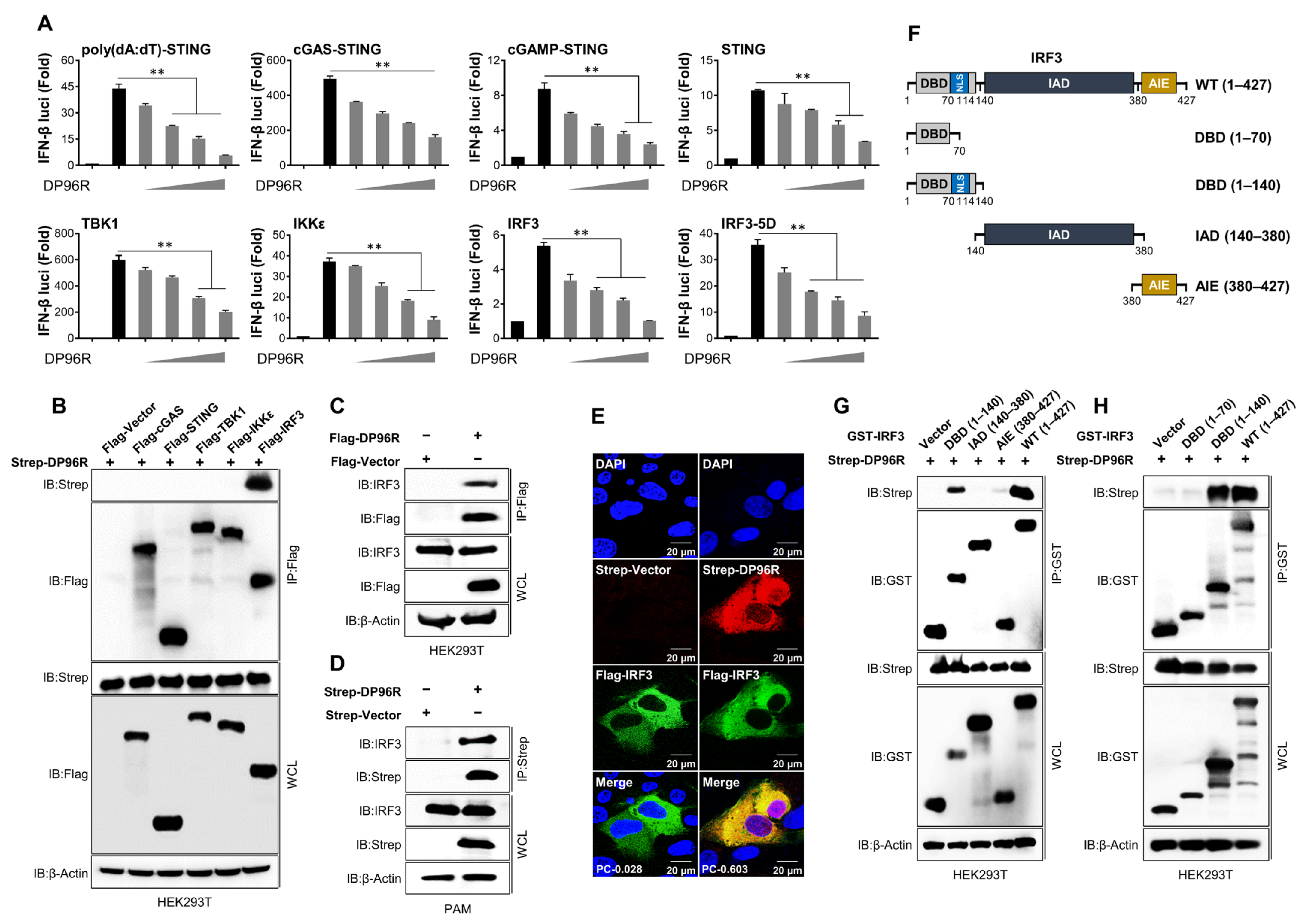
2.1. DP96R Targets IRF3
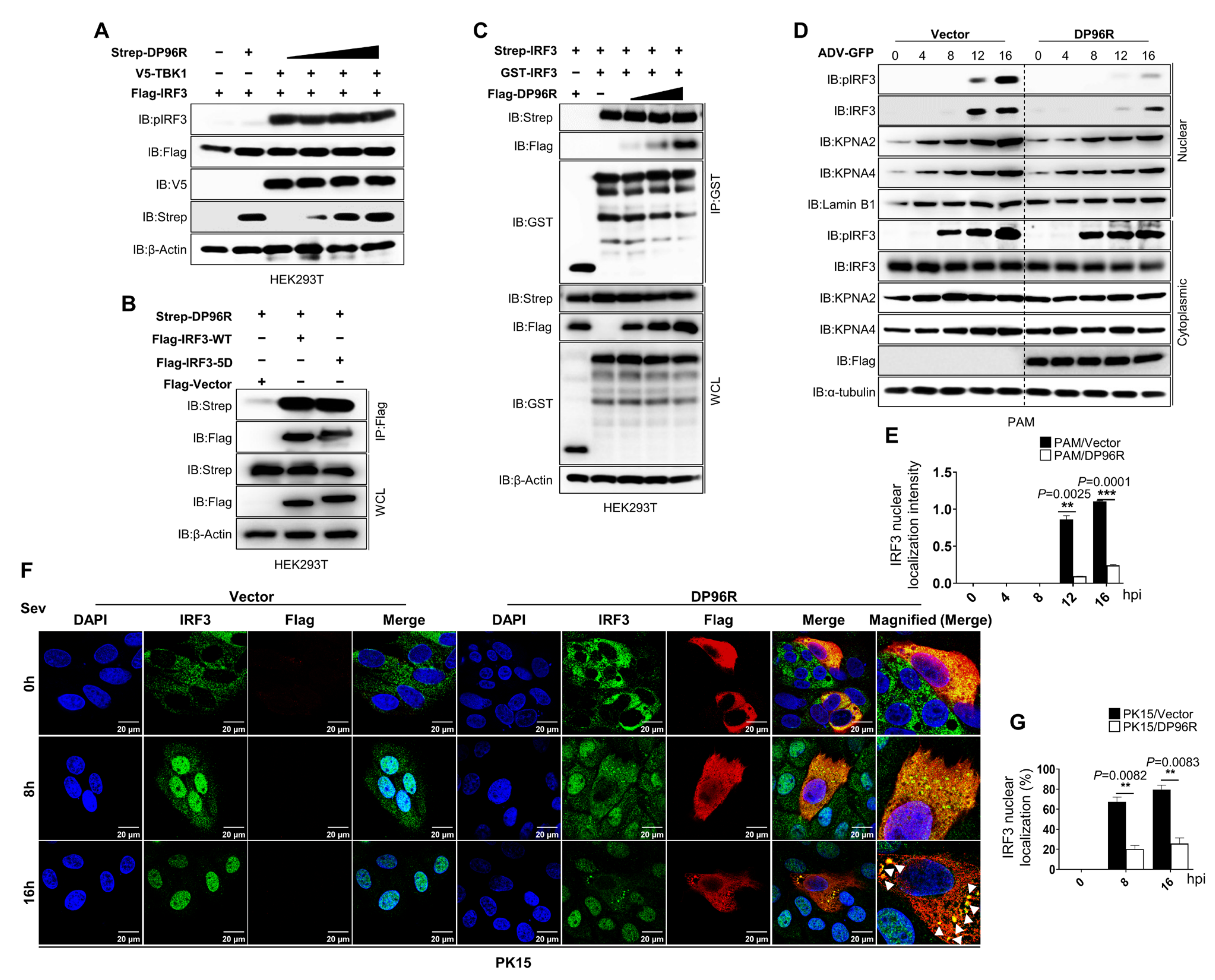
2.2. DP96R Impairs the Nuclear Translocation of IRF3
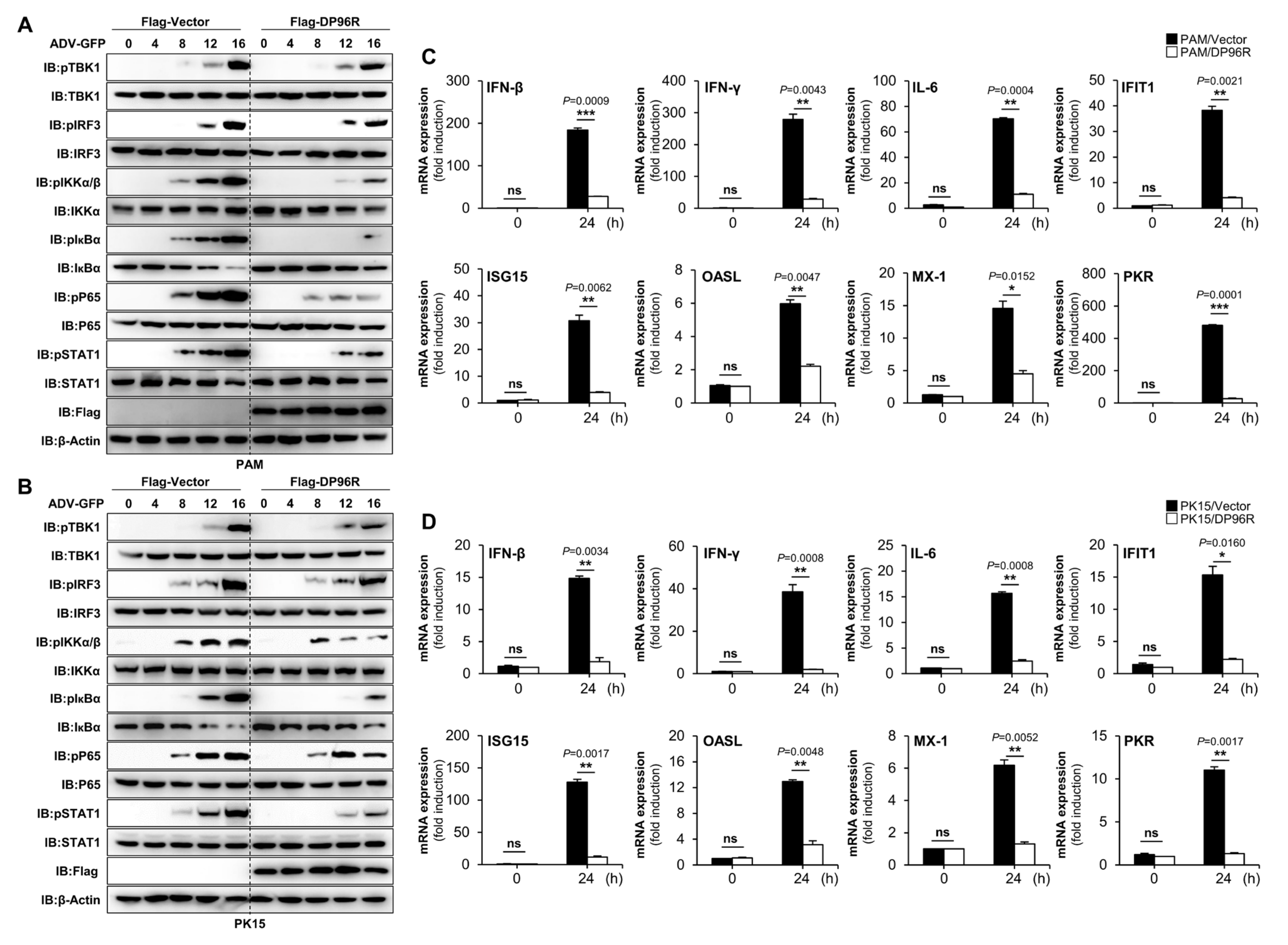
2.3. DP96R Disrupts Type I IFN Signaling and Subsequent Transcription of Antiviral Genes
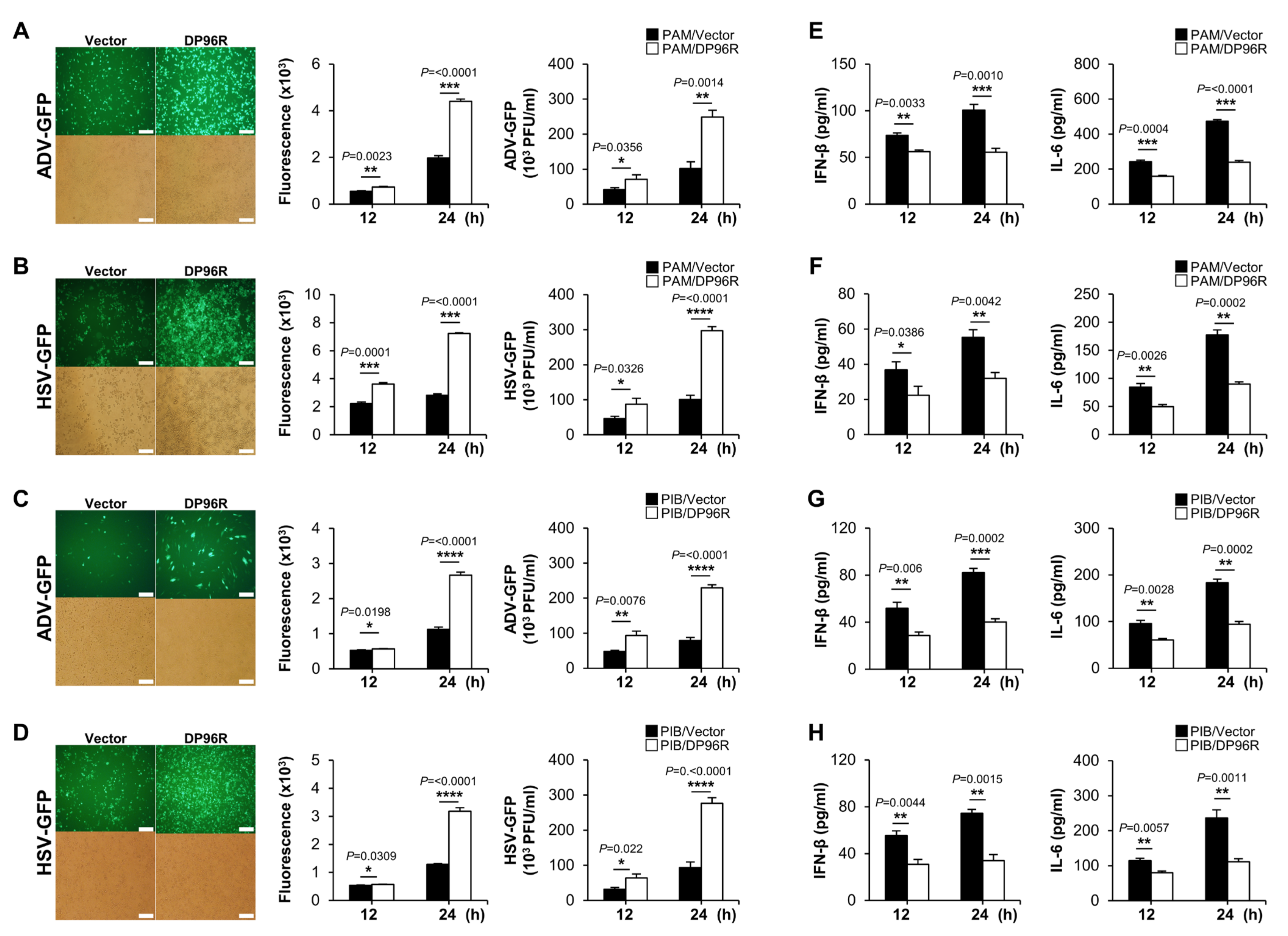
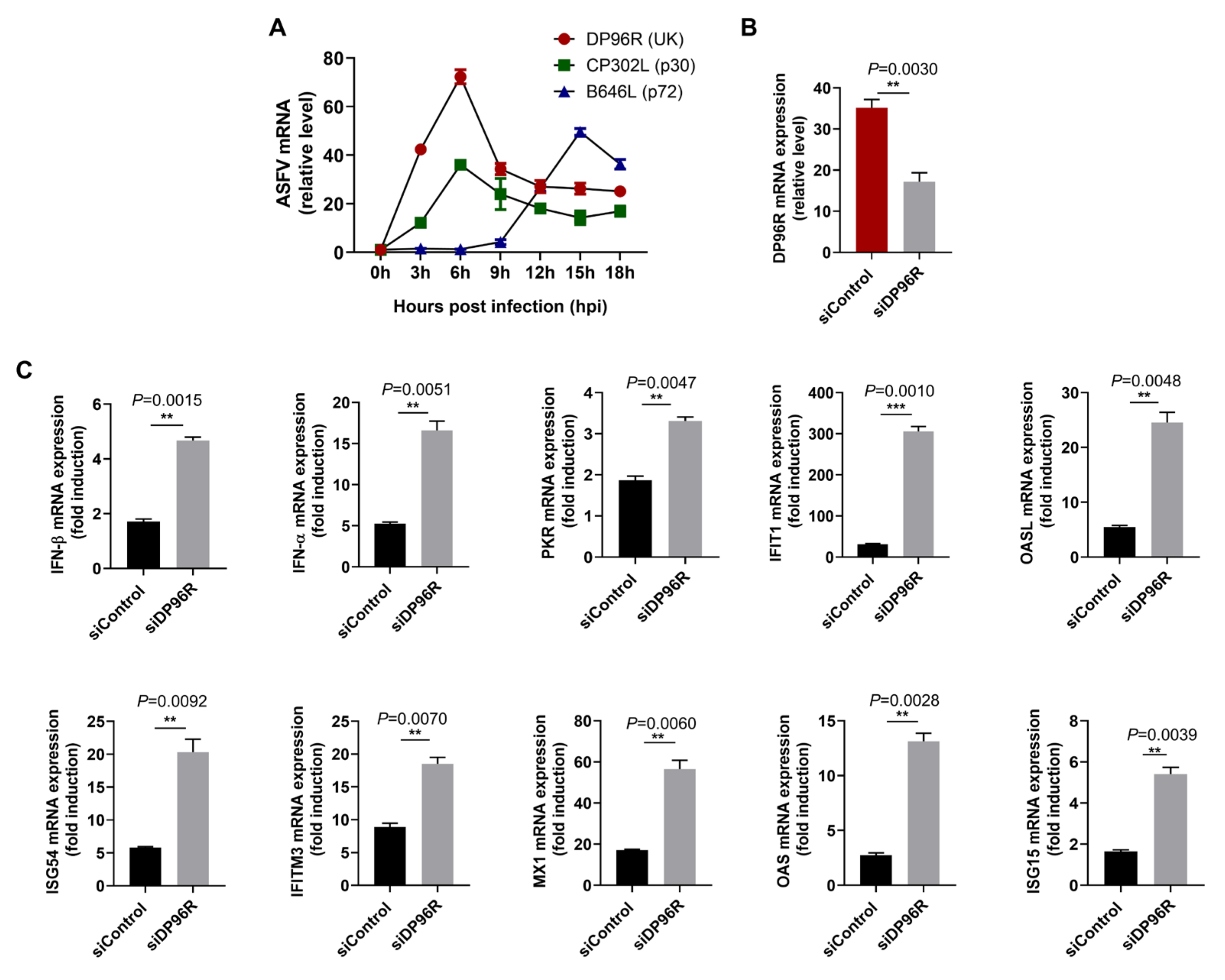
2.4. DP96R Negatively Regulates Innate Immune Responses against Viral Infection
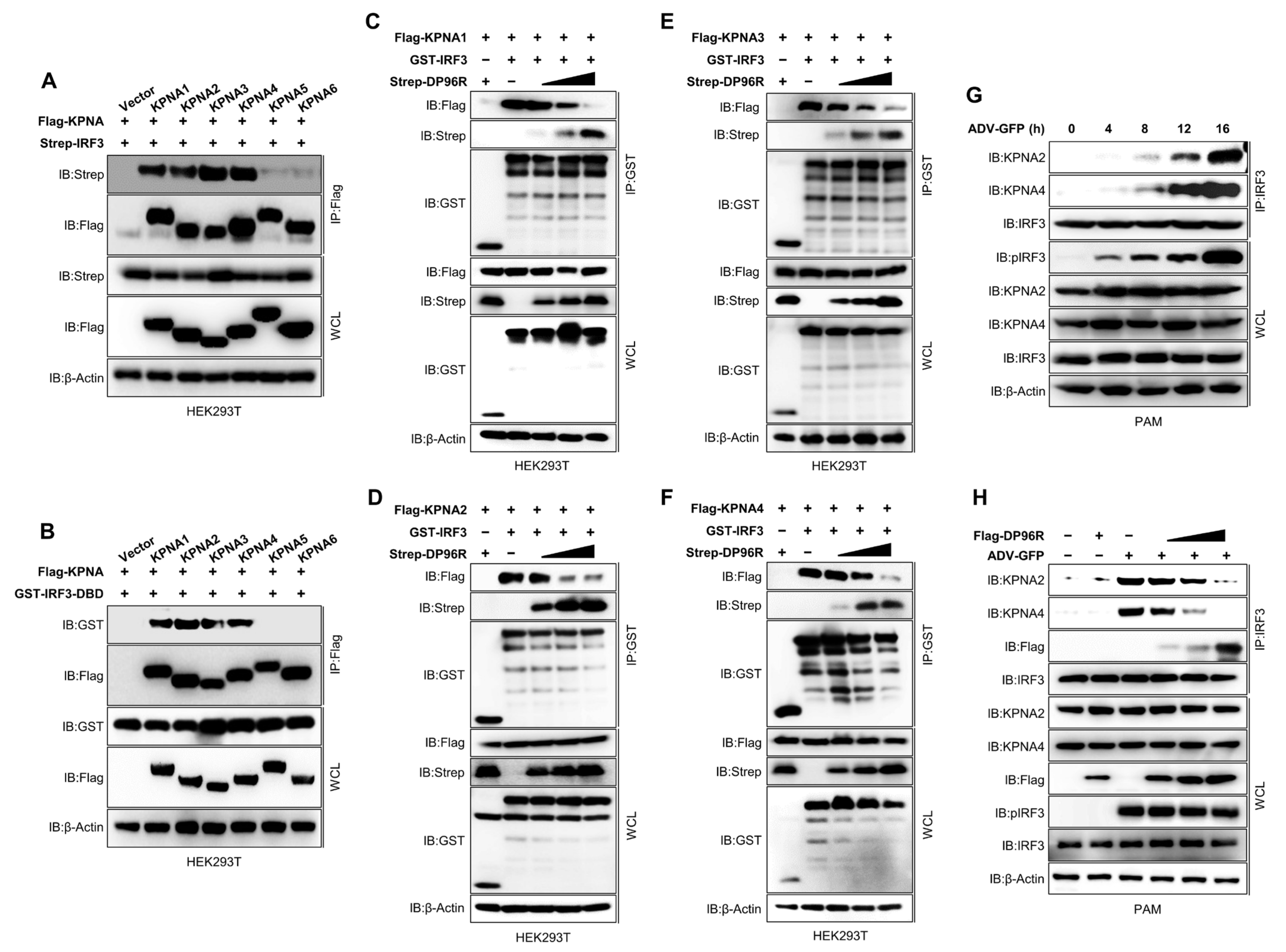
2.5. DP96R Inhibits Interaction between IRF3 and KPNA
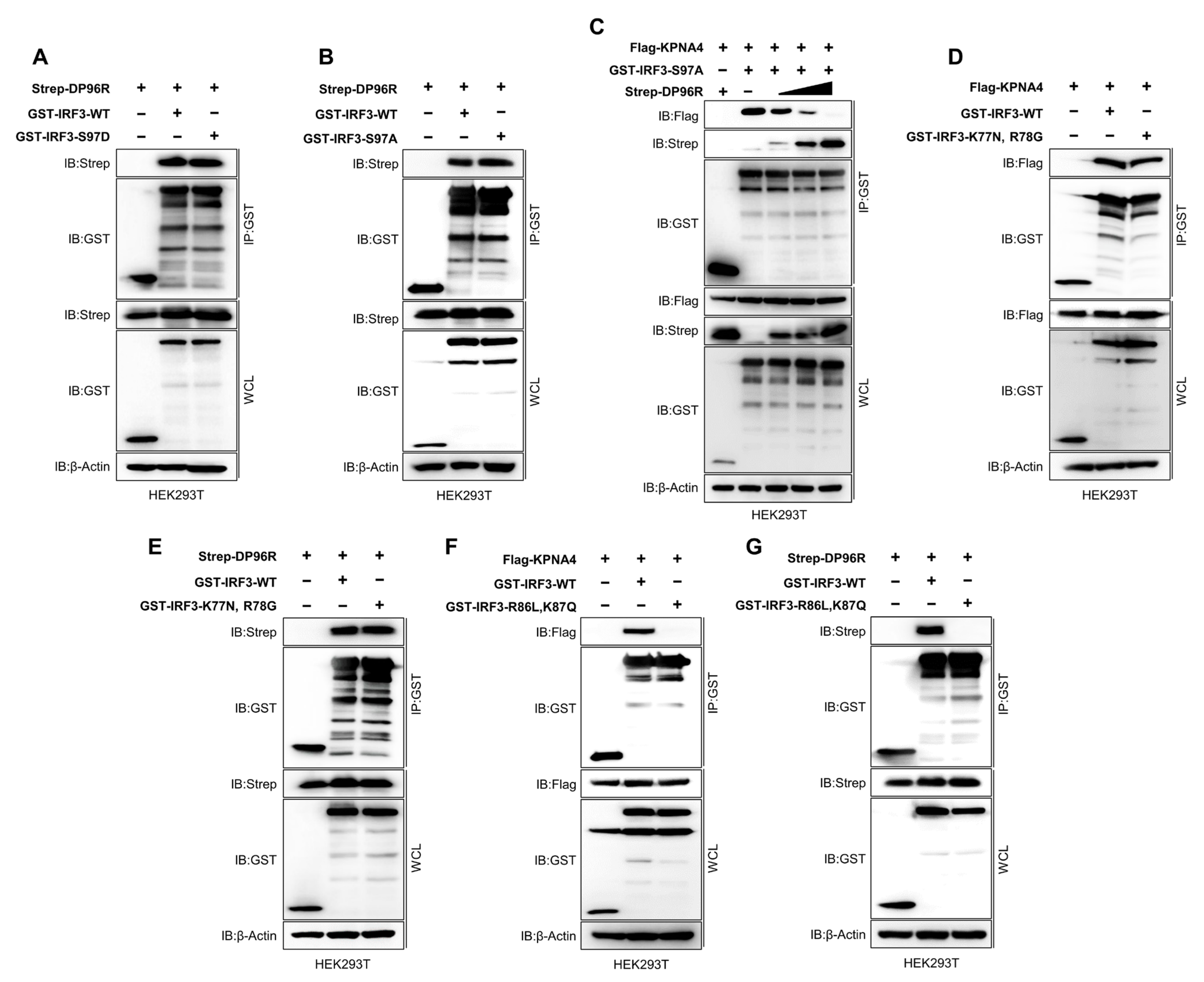
2.6. DP96R Interacts with the Major KPNA-Binding Site within IRF3
2.7. DP96R Is an Early Transcribed Protein Involved in the Antagonism of IFN and ISG Transcription
2.8. The Central Region of DP96R Regulates Immune Evasion
3. Discussion
4. Materials and Methods
4.1. Cells and Antibodies
4.2. Porcine Bone Marrow-Derived Macrophage Isolation
4.3. Porcine Bone Marrow-Derived Macrophage Immortalization
4.4. Continuous ASFV DP96R Protein-Expressing Cell Generation
4.5. Plasmids
4.6. Virus Infection and Plasmid Transfection
4.7. Virus Titration
4.8. DP96R Transcription Assay
4.9. RNA Interference Experiments
4.10. ELISA
4.11. Quantitative Real-Time PCR
4.12. Immunoprecipitation
4.13. Immunoblot Analysis
4.14. Luciferase Reporter Assay
4.15. Nuclear Fractionation Assay
4.16. Immunofluorescence and Confocal Microscopy
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dixon, L.K.; Sun, H.; Roberts, H. African swine fever. Antivir. Res. 2019, 165, 34–41. [Google Scholar] [CrossRef]
- Galindo, I.; Alonso, C. African Swine Fever Virus: A Review. Viruses 2017, 9, 103. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Madden, D.W.; Wilson, W.C.; Trujillo, J.D.; Richt, J.A. African Swine Fever Virus: An Emerging DNA Arbovirus. Front. Vet. Sci. 2020, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Borca, M.; Dixon, L.; Revilla, Y.; Rodriguez, F.; Escribano, J.M.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Asfarviridae. J. Gen. Virol. 2018, 99, 613–614. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, P. The persistence of African swine fever in Africa and the Mediterranean. Prev. Vet. Med. 1984, 2, 71–82. [Google Scholar] [CrossRef]
- Li, J.; Song, J.; Kang, L.; Huang, L.; Zhou, S.; Hu, L.; Zheng, J.; Li, C.; Zhang, X.; He, X. pMGF505-7R determines pathogenicity of African swine fever virus infection by inhibiting IL-1β and type I IFN production. PLoS Pathog. 2021, 17, e1009733. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cordón, P.; Montoya, M.; Reis, A.; Dixon, L. African swine fever: A re-emerging viral disease threatening the global pig industry. Vet. J. 2018, 233, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andrés, G. A Proteomic Atlas of the African Swine Fever Virus Particle. J. Virol. 2018, 92, e01293-18. [Google Scholar] [CrossRef] [PubMed]
- Karger, A.; Pérez-Núñez, D.; Urquiza, J.; Hinojar, P.; Alonso, C.; Freitas, F.B.; Revilla, Y.; Le Potier, M.F.; Montoya, M. An Update on African Swine Fever Virology. Viruses 2019, 11, 864. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Yu, L.; Liu, P. Cytosolic DNA sensing by cGAS: Regulation, function, and human diseases. Signal Transduct. Target. Ther. 2021, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Omura, H.; Ishitani, R.; Nureki, O. Cyclic GMP-AMP as an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Annu. Rev. Biochem. 2017, 86, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Ma, Z.; Damania, B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef]
- Deng, L.; Zeng, Q.; Wang, M.; Cheng, A.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Yang, Q.; Wu, Y. Suppression of NF-κB activity: A viral immune evasion mechanism. Viruses 2018, 10, 409. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Modulation of NF-κB signalling by microbial pathogens. Nat. Rev. Microbiol. 2011, 9, 291–306. [Google Scholar] [CrossRef]
- García-Belmonte, R.; Pérez-Núñez, D.; Pittau, M.; Richt, J.A.; Revilla, Y. African Swine Fever Virus Armenia/07 Virulent Strain Controls Interferon Beta Production through the cGAS-STING Pathway. J. Virol. 2019, 93, e02298-18. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, W.; Qiu, Z.; Li, Y.; Fan, J.; Wu, K.; Li, X.; Zhao, M.; Ding, H.; Fan, S. African Swine Fever Virus: A Review. Life 2022, 12, 1255. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, W.; Li, L.; Li, P.; Ma, Z.; Zhang, J.; Qi, X.; Ren, J.; Ru, Y.; Niu, Q. African swine fever virus MGF-505-7R negatively regulates cGAS–STING-mediated signaling pathway. J. Immunol. 2021, 206, 1844–1857. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Z.; Feng, T.; Ma, Z.; Xue, Q.; Wu, P.; Li, P.; Li, S.; Yang, F.; Cao, W. African swine fever virus E120R protein inhibits interferon beta production by interacting with IRF3 to block its activation. J. Virol. 2021, 95, e00824-21. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ao, D.; Jiang, S.; Xia, N.; Xu, Y.; Shao, Q.; Luo, J.; Wang, H.; Zheng, W.; Chen, N. African swine fever virus A528R inhibits TLR8 mediated NF-κB activity by targeting p65 activation and nuclear translocation. Viruses 2021, 13, 2046. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, S.; Xin, T.; Wang, X.; Yu, H.; Chen, S.; Jiang, Y.; Gao, X.; Jiang, Y.; Guo, X.; et al. African Swine Fever Virus MGF360-14L Negatively Regulates Type I Interferon Signaling by Targeting IRF3. Front. Cell Infect. Microbiol. 2021, 11, 818969. [Google Scholar] [CrossRef]
- Yang, J.; Li, S.; Feng, T.; Zhang, X.; Yang, F.; Cao, W.; Chen, H.; Liu, H.; Zhang, K.; Zhu, Z.; et al. African Swine Fever Virus F317L Protein Inhibits NF-κB Activation to Evade Host Immune Response and Promote Viral Replication. mSphere 2021, 6, e0065821. [Google Scholar] [CrossRef]
- Yang, K.; Huang, Q.; Wang, R.; Zeng, Y.; Cheng, M.; Xue, Y.; Shi, C.; Ye, L.; Yang, W.; Jiang, Y. African swine fever virus MGF505-11R inhibits type I interferon production by negatively regulating the cGAS-STING-mediated signaling pathway. Vet. Microbiol. 2021, 263, 109265. [Google Scholar] [CrossRef]
- Zhuo, Y.; Guo, Z.; Ba, T.; Zhang, C.; He, L.; Zeng, C.; Dai, H. African swine fever virus MGF360-12L inhibits type I interferon production by blocking the interaction of importin α and NF-κB signaling pathway. Virol. Sin. 2021, 36, 176–186. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Z.; Gao, X.; Lv, J.; Hu, Y.; Jung, Y.-S.; Zhu, S.; Wu, X.; Qian, Y.; Dai, J. ASFV pD345L protein negatively regulates NF-κB signalling by inhibiting IKK kinase activity. Vet. Res. 2022, 53, 32. [Google Scholar] [CrossRef]
- Cheng, M.; Luo, J.; Duan, Y.; Yang, Y.; Shi, C.; Sun, Y.; Lu, Y.; Wang, J.; Li, X.; Wang, J. African swine fever virus MGF505-3R inhibits cGAS-STING-mediated IFN-β pathway activation by degrading TBK1. Anim. Dis. 2022, 2, 13. [Google Scholar] [CrossRef]
- Dodantenna, N.; Ranathunga, L.; Chathuranga, W.G.; Weerawardhana, A.; Cha, J.-W.; Subasinghe, A.; Gamage, N.; Haluwana, D.; Kim, Y.; Jheong, W. African Swine Fever Virus EP364R and C129R Target Cyclic GMP-AMP To Inhibit the cGAS-STING Signaling Pathway. J. Virol. 2022, 96, e01022-22. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Yu, S.; Ge, H.; Wang, T.; Li, Y.; Zhou, P.; Pan, L.; Han, Y.; Yang, Y.; Sun, Y.; et al. The A137R Protein of African Swine Fever Virus Inhibits Type I Interferon Production via the Autophagy-Mediated Lysosomal Degradation of TBK1. J. Virol. 2022, 96, e0195721. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Xue, Y.; Niu, H.; Shi, C.; Cheng, M.; Wang, J.; Zou, B.; Wang, J.; Niu, T.; Bao, M. African swine fever virus MGF360-11L negatively regulates cGAS-STING-mediated inhibition of type I interferon production. Vet. Res. 2022, 53, 1–12. [Google Scholar] [CrossRef]
- Zsak, L.; Caler, E.; Lu, Z.; Kutish, G.; Neilan, J.; Rock, D. A nonessential African swine fever virus gene UK is a significant virulence determinant in domestic swine. J. Virol. 1998, 72, 1028–1035. [Google Scholar] [CrossRef]
- O’Donnell, V.; Risatti, G.R.; Holinka, L.G.; Krug, P.W.; Carlson, J.; Velazquez-Salinas, L.; Azzinaro, P.A.; Gladue, D.P.; Borca, M.V. Simultaneous deletion of the 9GL and UK genes from the African swine fever virus Georgia 2007 isolate offers increased safety and protection against homologous challenge. J. Virol. 2017, 91, e01760-16. [Google Scholar] [CrossRef] [PubMed]
- Teklue, T.; Wang, T.; Luo, Y.; Hu, R.; Sun, Y.; Qiu, H.-J. Generation and evaluation of an African swine fever virus mutant with deletion of the CD2v and UK genes. Vaccines 2020, 8, 763. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, D.; He, X.; Liu, R.; Wang, Z.; Zhang, X.; Li, F.; Shan, D.; Chen, H.; Zhang, J. A seven-gene-deleted African swine fever virus is safe and effective as a live attenuated vaccine in pigs. Sci. China Life Sci. 2020, 63, 623–634. [Google Scholar] [CrossRef]
- Zhu, M.; Fang, T.; Li, S.; Meng, K.; Guo, D. Bipartite nuclear localization signal controls nuclear import and DNA-binding activity of IFN regulatory factor 3. J. Immunol. 2015, 195, 289–297. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.K.; Kumar, K.P.; Reich, N.C. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol. Cell Biol. 1998, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Holly, M.K.; Smith, J.G. Adenovirus infection of human enteroids reveals interferon sensitivity and preferential infection of goblet cells. J. Virol. 2018, 92, e00250-18. [Google Scholar] [CrossRef] [PubMed]
- Altinkilic, B.; Brandner, G. Interferon inhibits herpes simplex virus-specific translation: A reinvestigation. J. Gen. Virol. 1988, 69, 3107–3112. [Google Scholar] [CrossRef]
- Stewart, W.E.; Scott, W.D.; Sulkin, S.E. Relative sensitivities of viruses to different species of interferon. J. Virol. 1969, 4, 147–153. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, Y.E.; Palazzo, A.F. Crosstalk between nucleocytoplasmic trafficking and the innate immune response to viral infection. J. Biol. Chem. 2021, 297, 100856. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Glanz, A.; Chakravarty, S.; Varghese, M.; Kottapalli, A.; Fan, S.; Chakravarti, R.; Chattopadhyay, S. Transcriptional and non-transcriptional activation, posttranslational modifications, and antiviral functions of interferon regulatory factor 3 and viral antagonism by the SARS-coronavirus. Viruses 2021, 13, 575. [Google Scholar] [CrossRef]
- Görlich, D.; Kraft, R.; Kostka, S.; Vogel, F.; Hartmann, E.; Laskey, R.A.; Mattaj, I.W.; Izaurralde, E. Importin provides a link between nuclear protein import and U snRNA export. Cell 1996, 87, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Nucleocytoplasmic transport: Signals, mechanisms and regulation. Nature 1997, 386, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Chen, Z.; Li, Y.; Zhao, Z.; He, W.; Zohaib, A.; Song, Y.; Deng, C.; Zhang, B.; Chen, H. Japanese encephalitis virus NS5 inhibits type I interferon (IFN) production by blocking the nuclear translocation of IFN regulatory factor 3 and NF-κB. J. Virol. 2017, 91, e00039-17. [Google Scholar] [CrossRef]
- Khan, H.; Sumner, R.P.; Rasaiyaah, J.; Tan, C.P.; Rodriguez-Plata, M.T.; Van Tulleken, C.; Fink, D.; Zuliani-Alvarez, L.; Thorne, L.; Stirling, D. HIV-1 Vpr antagonizes innate immune activation by targeting karyopherin-mediated NF-κB/IRF3 nuclear transport. Elife 2020, 9, e60821. [Google Scholar] [CrossRef]
- Kumar, K.P.; McBride, K.M.; Weaver, B.K.; Dingwall, C.; Reich, N.C. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell. Biol. 2000, 20, 4159–4168. [Google Scholar] [CrossRef]
- Li, J.; Lu, M.; Huang, B.; Lv, Y. Porcine circovirus type 2 inhibits interferon-β expression by targeting Karyopherin alpha-3 in PK-15 cells. Virology 2018, 520, 75–82. [Google Scholar] [CrossRef]
- Zhang, L.; Qiu, S.; Lu, M.; Huang, C.; Lv, Y. Nuclear transporter karyopherin subunit alpha 3 levels modulate Porcine circovirus type 2 replication in PK-15ácells. Virology 2020, 548, 31–38. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, M.-X.; Tang, Z.; Zhang, Q.; Ye, J.; Xiong, T.-C.; Zhang, Z.-D.; Zhong, B. USP22 promotes IRF3 nuclear translocation and antiviral responses by deubiquitinating the importin protein KPNA2. J. Exp. Med. 2020, 217, e20191174. [Google Scholar] [CrossRef]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.-Y.; Chen, S.; Zhao, X.; Lei, C.-Q.; Guo, L.; Chen, Y. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ke, J.; Zhang, J.; Yang, J.; Yue, H.; Zhou, X.; Qi, Y.; Zhu, R.; Miao, F.; Li, Q. African swine fever virus bearing an I226R gene deletion elicits robust immunity in pigs to African swine fever. J. Virol. 2021, 95, e02185-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, J.; Wu, Y.; Chen, H.; Zhang, S.; Li, J.; Xin, T.; Jia, H.; Hou, S.; Jiang, Y. Inhibition of cGAS-STING-TBK1 signaling pathway by DP96R of ASFV China 2018/1. Biochem. Biophys. Res. Commun. 2018, 506, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.; Piccone, M.; Zaffuto, K.; Neilan, J.; Kutish, G.; Lu, Z.; Balinsky, C.; Gibb, T.; Bean, T.; Zsak, L. African swine fever virus multigene family 360 and 530 genes affect host interferon response. J. Virol. 2004, 78, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.L.; Abrams, C.C.; Goatley, L.C.; Netherton, C.; Chapman, D.G.; Sanchez-Cordon, P.; Dixon, L.K. Deletion of African swine fever virus interferon inhibitors from the genome of a virulent isolate reduces virulence in domestic pigs and induces a protective response. Vaccine 2016, 34, 4698–4705. [Google Scholar] [CrossRef]
- Huang, L.; Chen, W.; Liu, H.; Xue, M.; Dong, S.; Liu, X.; Feng, C.; Cao, S.; Ye, G.; Zhou, Q. African Swine Fever Virus HLJ/18 CD2v Suppresses Type I IFN Production and IFN-Stimulated Genes Expression through Negatively Regulating cGMP-AMP Synthase–STING and IFN Signaling Pathways. J. Immunol. 2023, 210, 1338–1350. [Google Scholar] [CrossRef]
- Ran, Y.; Li, D.; Xiong, M.G.; Liu, H.N.; Feng, T.; Shi, Z.W.; Li, Y.H.; Wu, H.N.; Wang, S.Y.; Zheng, H.X.; et al. African swine fever virus I267L acts as an important virulence factor by inhibiting RNA polymerase III-RIG-I-mediated innate immunity. PLoS Pathog. 2022, 18, e1010270. [Google Scholar] [CrossRef]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623. [Google Scholar] [CrossRef]
- Lohoff, M.; Mak, T.W. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nat. Rev. Immunol. 2005, 5, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Chiba, S.; Hangai, S.; Kometani, K.; Inoue, A.; Kimura, Y.; Abe, T.; Kiyonari, H.; Nishio, J.; Taguchi-Atarashi, N. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 5253–5258. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; TenOever, B.R.; Grandvaux, N.; Zhou, G.-P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar] [CrossRef] [PubMed]
- Chathuranga, K.; Weerawardhana, A.; Dodantenna, N.; Lee, J.-S. Regulation of antiviral innate immune signaling and viral evasion following viral genome sensing. Exp. Mol. Med. 2021, 53, 1647–1668. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Paterson, R.G.; Stock, N.; Durbin, J.E.; Durbin, R.K.; Goodbourn, S.; Randall, R.E.; Lamb, R.A. Recovery of paramyxovirus simian virus 5 with a V protein lacking the conserved cysteine-rich domain: The multifunctional V protein blocks both interferon-β induction and interferon signaling. Virology 2002, 303, 15–32. [Google Scholar] [CrossRef]
- Donelan, N.R.; Dauber, B.; Wang, X.; Basler, C.F.; Wolff, T.; García-Sastre, A. The N-and C-terminal domains of the NS1 protein of influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J. Virol. 2004, 78, 11574–11582. [Google Scholar] [CrossRef]
- Delhaye, S.; Van Pesch, V.; Michiels, T. The leader protein of Theiler’s virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 2004, 78, 4357–4362. [Google Scholar] [CrossRef]
- Paladino, P.; Collins, S.E.; Mossman, K.L. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS ONE 2010, 5, e10428. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell. Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef]
- Fung, S.-Y.; Siu, K.-L.; Lin, H.; Yeung, M.L.; Jin, D.-Y. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int. J. Biol. Sci. 2021, 17, 1547. [Google Scholar] [CrossRef]
- Oka, M.; Yoneda, Y. Importin α: Functions as a nuclear transport factor and beyond. Proc. Jpn. Acad. Ser. B 2018, 94, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, R.; Fairbairn, L.; Beraldi, D.; Sester, D.P.; Archibald, A.L.; Tuggle, C.K.; Hume, D.A. Pig bone marrow-derived macrophages resemble human macrophages in their response to bacterial lipopolysaccharide. J. Immunol. 2012, 188, 3382–3394. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, R.; Fairbairn, L.; Downing, A.; Beraldi, D.; Sester, D.P.; Freeman, T.C.; Tuggle, C.K.; Archibald, A.L.; Hume, D.A. The impact of breed and tissue compartment on the response of pig macrophages to lipopolysaccharide. BMC Genom. 2013, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Scheenstra, M.R.; van Dijk, A.; Veldhuizen, E.J.; Haagsman, H.P. A new and efficient culture method for porcine bone marrow-derived M1-and M2-polarized macrophages. Vet. Immunol. Immunopathol. 2018, 200, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Kim, T.-H.; Lee, H.-C.; Nikapitiya, C.; Uddin, M.B.; Park, M.-E.; Pathinayake, P.; Lee, E.S.; Chathuranga, K.; Herath, T.U. Rubicon modulates antiviral type I interferon (IFN) signaling by targeting IFN regulatory factor 3 dimerization. J. Virol. 2017, 91, e00248-17. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dodantenna, N.; Cha, J.-W.; Chathuranga, K.; Chathuranga, W.A.G.; Weerawardhana, A.; Ranathunga, L.; Kim, Y.; Jheong, W.; Lee, J.-S. The African Swine Fever Virus Virulence Determinant DP96R Suppresses Type I IFN Production Targeting IRF3. Int. J. Mol. Sci. 2024, 25, 2099. https://doi.org/10.3390/ijms25042099
Dodantenna N, Cha J-W, Chathuranga K, Chathuranga WAG, Weerawardhana A, Ranathunga L, Kim Y, Jheong W, Lee J-S. The African Swine Fever Virus Virulence Determinant DP96R Suppresses Type I IFN Production Targeting IRF3. International Journal of Molecular Sciences. 2024; 25(4):2099. https://doi.org/10.3390/ijms25042099
Chicago/Turabian StyleDodantenna, Niranjan, Ji-Won Cha, Kiramage Chathuranga, W. A. Gayan Chathuranga, Asela Weerawardhana, Lakmal Ranathunga, Yongkwan Kim, Weonhwa Jheong, and Jong-Soo Lee. 2024. "The African Swine Fever Virus Virulence Determinant DP96R Suppresses Type I IFN Production Targeting IRF3" International Journal of Molecular Sciences 25, no. 4: 2099. https://doi.org/10.3390/ijms25042099
APA StyleDodantenna, N., Cha, J. -W., Chathuranga, K., Chathuranga, W. A. G., Weerawardhana, A., Ranathunga, L., Kim, Y., Jheong, W., & Lee, J. -S. (2024). The African Swine Fever Virus Virulence Determinant DP96R Suppresses Type I IFN Production Targeting IRF3. International Journal of Molecular Sciences, 25(4), 2099. https://doi.org/10.3390/ijms25042099