

Mesenchymal Transglutaminase 2 Activates Epithelial ADAM17: Link to G-Protein-Coupled Receptor 56 (ADGRG1) Signalling

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
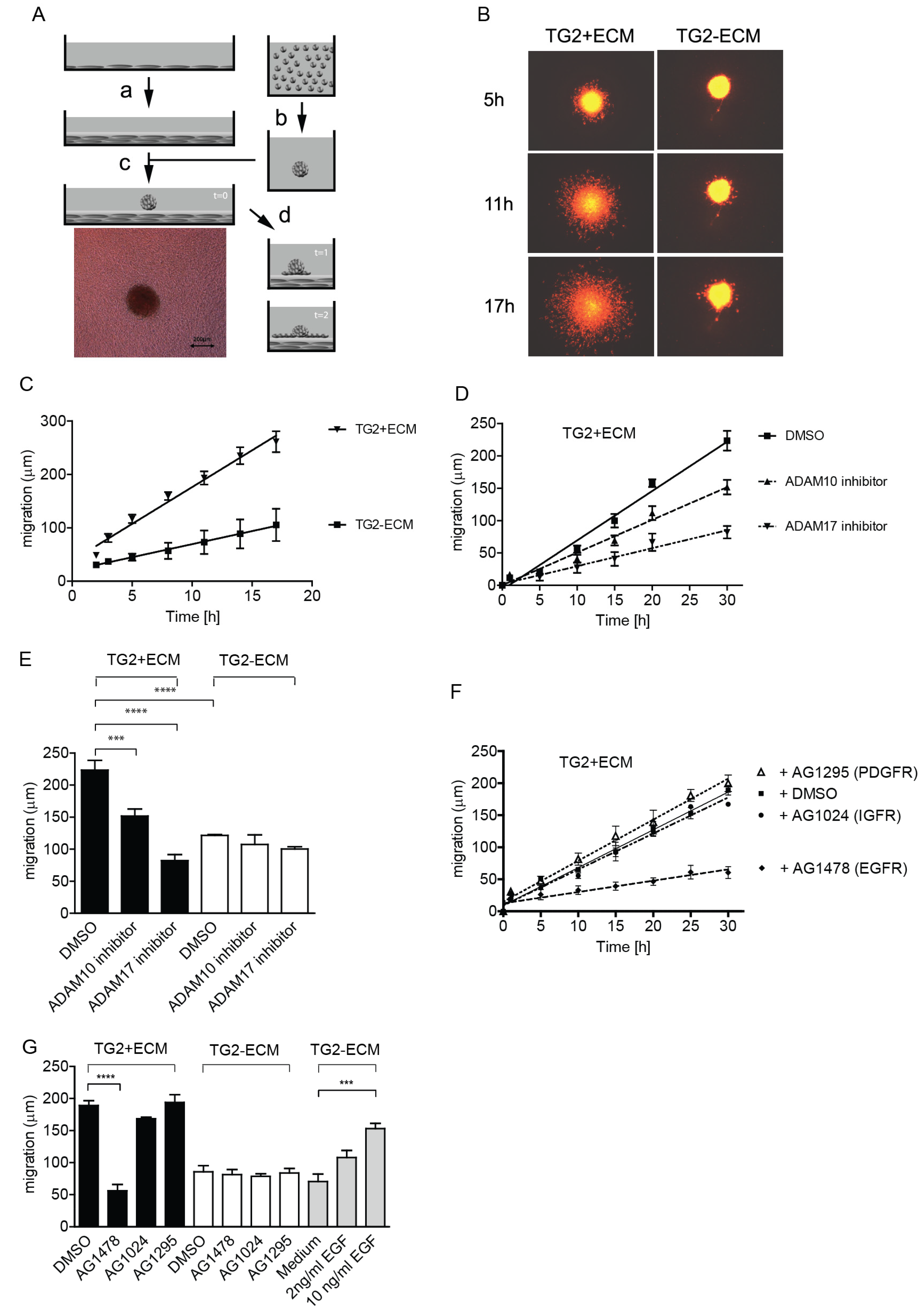
2.1. TG2-Positive ECM Facilitates Keratinocyte Re-Epithelialisation Response through ADAM17-Dependent EGFR Transactivation
2.2. TG2 Promotes Keratinocyte Proliferation through ADAM17 Dependent EGFR-Ligand Shedding
2.3. GPR56 Signaling Regulates ADAM17 Activity
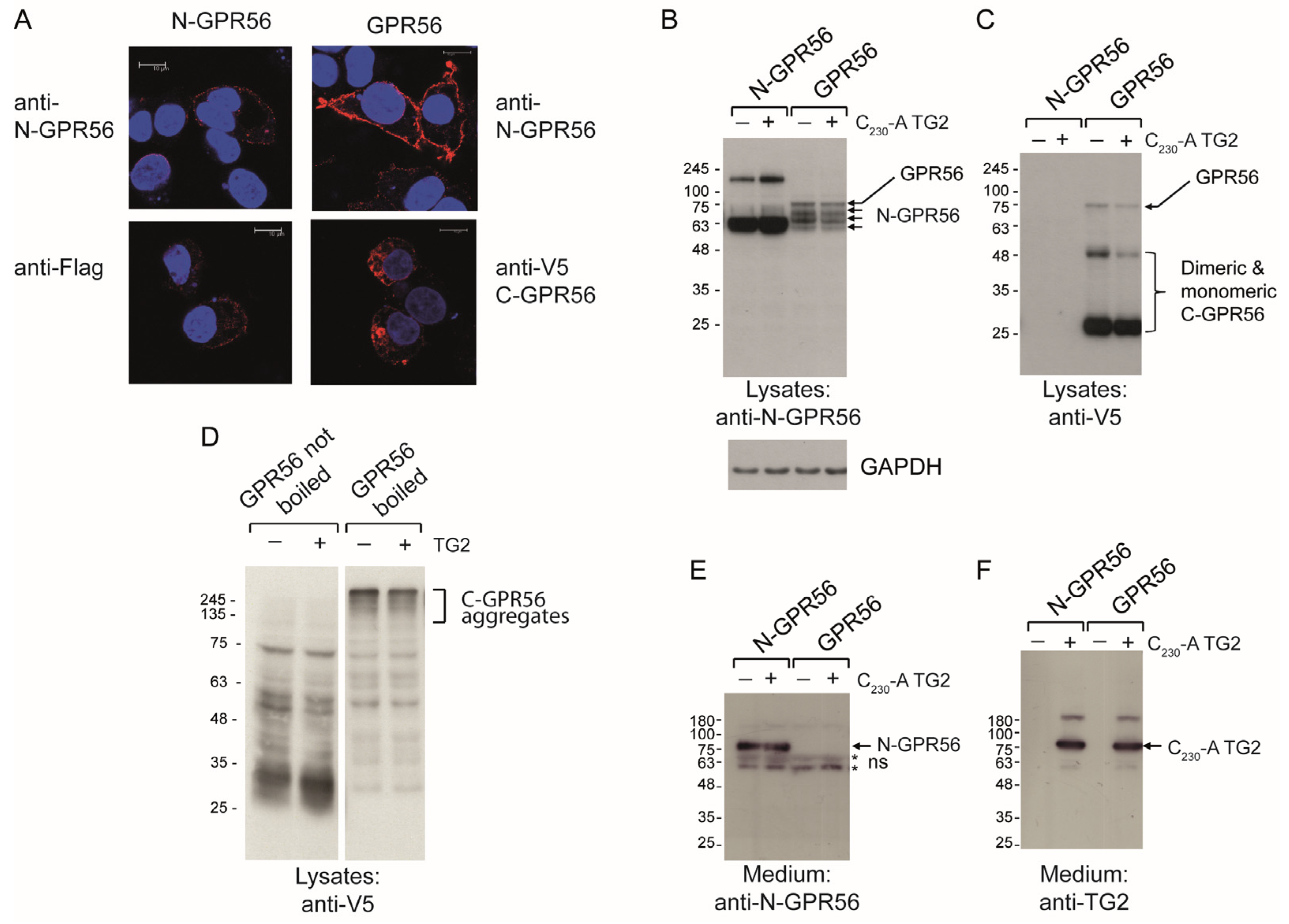
2.4. TG2 Treatment Does Not Induce Detectable Intermolecular Crosslinking of GPR56
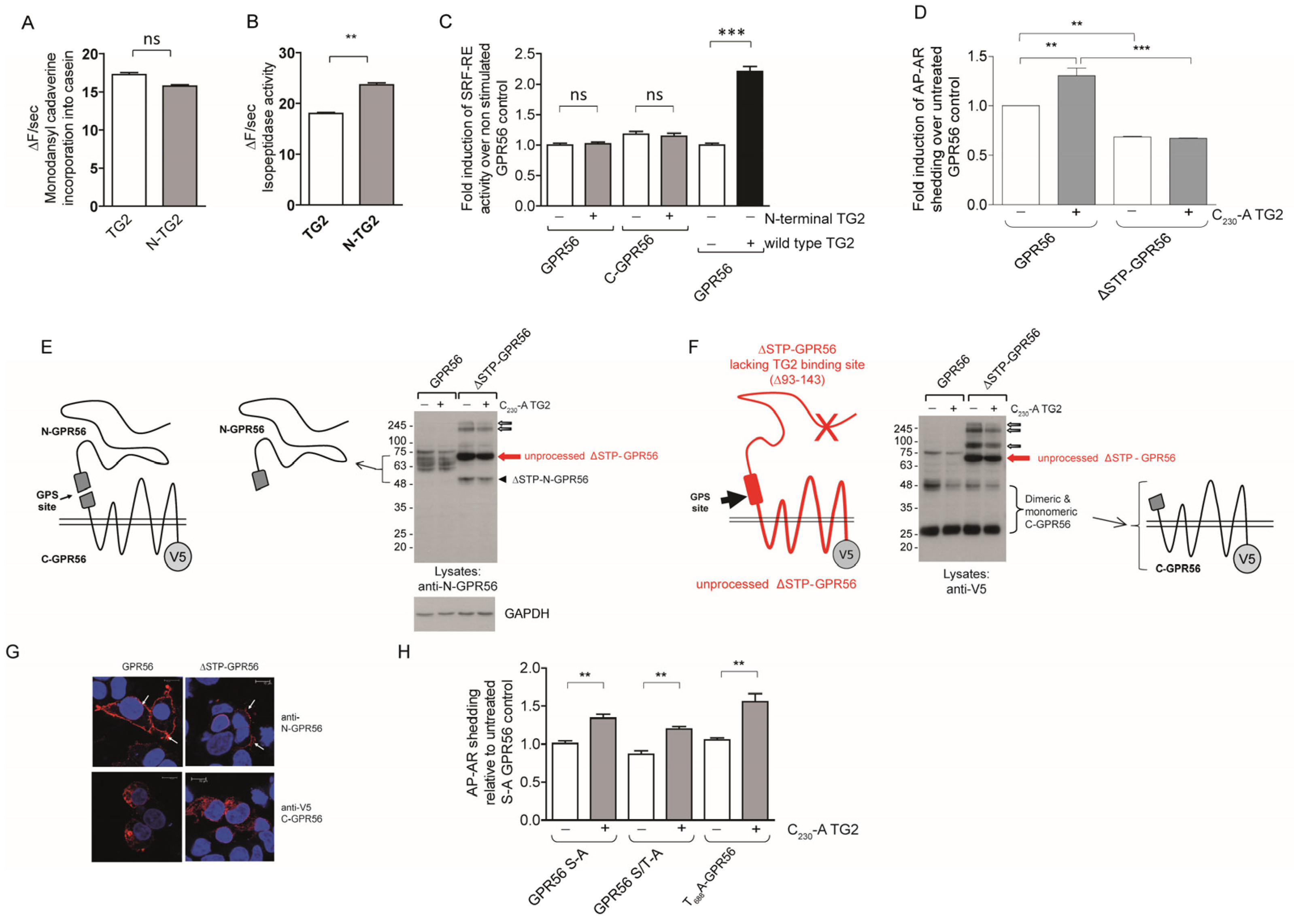
2.5. GPR56 Activation by TG2 or Agonistic N-GPR56 Antibody Requires N-Terminal GPR56 as Determined Using Shedding or Rho-Luciferase Reporter Assays
2.6. TG2-Dependent GPR56 Signaling Requires C-Terminal TG2 β-Barrels, but Is Independent of GPR56 Tail Phosphorylation Sites
2.7. TG2 Is Internalized Selectively in GPR56 Expressing Cells
3. Discussion
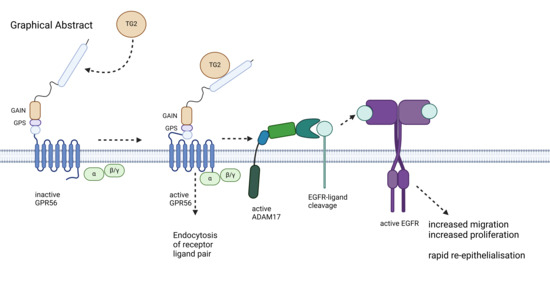
3.1. ADAM17 Activation by GPR56 and TG2 Establishes a Ligand Receptor Pair
3.2. GPR56 Domain Requirements for Agonist-Mediated ADAM17 Activation
3.3. GPR56 Is Required for Rapid Endocytosis of TG2, Leading to Partial Co-Localisation of TG2 and N-GPR56 in Endocytic Vesicles
3.4. TG2-Dependent Re-Epithelialisation Requires ADAM17-Dependent EGFR Transactivation
4. Material and Methods
4.1. Chemicals and Antibodies
4.2. Re-Epithelialisation Model
4.2.1. Production of TG2+ and TG2− ECM
4.2.2. Preparation of Fluorescently Labelled Keratinocyte Spheroids
4.2.3. Migration Studies on TG2+ and TG2− ECM Using Fluorescently Labelled N-Tert1 Keratinocytes Spheroids by Time Lapse Microscopy
4.2.4. Expression Constructs and Characterization of TG2, C230-A TG2, C277-S TG2, and N-Terminal TG2
Expression Constructs for N-GPR56-Fc Fusion Proteins
Expression of N-GPR56-Fc Fusion Proteins in CHO Cells
Purification of N-GPR56-Fc Fusion Proteins
TG2 Pull Down Assay
Surface Plasmon Resonance Analysis
4.2.5. Keratinocyte Proliferation
4.2.6. GPR56-Expression Analysis
4.2.7. GPR56-Expression Constructs
4.2.8. GPR56-Dependent Activation of ADAM17
4.2.9. Luciferase Reporter Assays
4.2.10. SDS-Page and Western Blotting
4.2.11. Immuno-Localisation of GPR56 in Transiently Transfected Cells
4.2.12. Internalisation of TG2 and SNAP-GPR56
4.2.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hakkinen, L.; Uitto, V.J.; Larjava, H. Cell biology of gingival wound healing. Periodontology 2000, 24, 127–152. [Google Scholar] [CrossRef]
- Müller, A.-K.; Meyer, M.; Werner, S. The roles of receptor tyrosine kinases and their ligands in the wound repair process. Semin. Cell Dev. Biol. 2012, 23, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Pilcher, B.K.; Dumin, J.; Schwartz, M.J.; Mast, B.A.; Schultz, G.S.; Parks, W.C.; Welgus, H.G. Keratinocyte collagenase-1 expression requires an epidermal growth factor receptor autocrine mechanism. J. Biol. Chem. 1999, 274, 10372–10381. [Google Scholar] [CrossRef] [PubMed]
- Franzke, C.-W.; Cobzaru, C.; Triantafyllopoulou, A.; Löffek, S.; Horiuchi, K.; Threadgill, D.W.; Kurz, T.; van Rooijen, N.; Bruckner-Tuderman, L.; Blobel, C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand–dependent terminal keratinocyte differentiation. J. Exp. Med. 2012, 209, 1105–1119. [Google Scholar] [CrossRef]
- Stephens, P.; Grenard, P.; Aeschlimann, P.; Langley, M.; Blain, E.; Errington, R.; Kipling, D.; Thomas, D.; Aeschlimann, D. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 2004, 117, 3389–3403. [Google Scholar] [CrossRef]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef]
- Szondy, Z.; Sarang, Z.; Molnár, P.; Németh, T.; Piacentini, M.; Mastroberardino, P.G.; Falasca, L.; Aeschlimann, D.; Kovács, J.; Kiss, I.; et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7812–7817. [Google Scholar] [CrossRef]
- Tong, L.; Png, E.; AiHua, H.; Yong, S.S.; Yeo, H.L.; Riau, A.; Mendoz, E.; Chaurasia, S.S.; Lim, C.T.; Yiu, T.W.; et al. Molecular mechanism of transglutaminase-2 in corneal epithelial migration and adhesion. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 1304–1315. [Google Scholar] [CrossRef]
- Oh, K.; Seo, M.W.; Lee, G.Y.; Byoun, O.-J.; Kang, H.-R.; Cho, S.-H.; Lee, D.-S. Airway epithelial cells initiate the allergen response through transglutaminase 2 by inducing IL-33 expression and a subsequent Th2 response. Respir. Res. 2013, 14, 35. [Google Scholar] [CrossRef]
- Sun, H.; Kaartinen, M.T. Transglutaminase activity regulates differentiation, migration and fusion of osteoclasts via affecting actin dynamics. J. Cell. Physiol. 2018, 233, 7497–7513. [Google Scholar] [CrossRef]
- Xu, L.; Begum, S.; Hearn, J.D.; Hynes, R.O. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 9023–9028. [Google Scholar] [CrossRef]
- Ganesh, R.A.; Sonpatki, P.; Naik, D.; John, A.E.; Sathe, G.; Lakshmikantha, A.; Chandrachari, K.P.; Bauer, L.; Knäuper, V.; Aeschlimann, D.; et al. Multi-Omics Analysis of Glioblastoma and Glioblastoma Cell Line: Molecular Insights Into the Functional Role of GPR56 and TG2 in Mesenchymal Transition. Front. Oncol. 2022, 12, 841890. [Google Scholar] [CrossRef]
- Borgatti, R.; Marelli, S.; Bernardini, L.; Novelli, A.; Cavallini, A.; Tonelli, A.; Bassi, M.T.; Dallapiccola, B. Bilateral frontoparietal polymicrogyria (BFPP) syndrome secondary to a 16q12.1-q21 chromosome deletion involving GPR56 gene. Clin. Genet. 2009, 76, 573–576. [Google Scholar] [CrossRef]
- Chiang, N.-Y.; Hsiao, C.-C.; Huang, Y.-S.; Chen, H.-Y.; Hsieh, I.-J.; Chang, G.-W.; Lin, H.-H. Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. J. Biol. Chem. 2011, 286, 14215–14225. [Google Scholar] [CrossRef]
- Ke, N.; Ma, H.; Diedrich, G.; Chionis, J.; Liu, G.; Yu, D.-H.; Wong-Staal, F.; Li, Q.-X. Biochemical characterization of genetic mutations of GPR56 in patients with bilateral frontoparietal polymicrogyria (BFPP). Biochem. Biophys. Res. Commun. 2008, 366, 314–320. [Google Scholar] [CrossRef]
- Luo, R.; Yang, H.M.; Jin, Z.; Halley, D.J.; Chang, B.S.; MacPherson, L.; Brueton, L.; Piao, X. A novel GPR56 mutation causes bilateral frontoparietal polymicrogyria. Pediatr. Neurol. 2011, 45, 49–53. [Google Scholar] [CrossRef]
- Ackerman, S.D.; Garcia, C.; Piao, X.; Gutmann, D.H.; Monk, K.R. The adhesion GPCR Gpr56 regulates oligodendrocyte development via interactions with Gα12/13 and RhoA. Nat. Commun. 2015, 6, 6122. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, S.; Luo, R.; Poitelon, Y.; Mogha, A.; Harty, B.L.; D’rozario, M.; Sanchez, N.E.; Lakkaraju, A.; Gamble, P.; Li, J.; et al. GPR56/ADGRG1 regulates development and maintenance of peripheral myelin. J. Exp. Med. 2018, 215, 941–961. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, T.; Sakata, K.; Yoshizaki, K.; Tago, K.; Mizuno, N.; Itoh, H. Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a Gα12/13 and Rho pathway. J. Biol. Chem. 2008, 283, 14469–14478. [Google Scholar] [CrossRef] [PubMed]
- Paavola, K.J.; Stephenson, J.R.; Ritter, S.L.; Alter, S.P.; Hall, R.A. The N terminus of the adhesion G protein-coupled receptor GPR56 controls receptor signaling activity. J. Biol. Chem. 2011, 286, 28914–28921. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Jeong, S.-J.; Jin, Z.; Strokes, N.; Li, S.; Piao, X. G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc. Natl. Acad. Sci. USA 2011, 108, 12925–12930. [Google Scholar] [CrossRef] [PubMed]
- Giera, S.; Luo, R.; Ying, Y.; Ackerman, S.D.; Jeong, S.-J.; Stoveken, H.M.; Folts, C.J.; Welsh, C.A.; Tall, G.G.; Stevens, B.; et al. Microglial transglutaminase-2 drives myelination and myelin repair via GPR56/ADGRG1 in oligodendrocyte precursor cells. eLife 2018, 7, e33385. [Google Scholar] [CrossRef] [PubMed]
- Araç, D.; Boucard, A.A.; Bolliger, M.F.; Nguyen, J.; Soltis, S.M.; Südhof, T.C.; Brunger, A.T. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 2012, 31, 1364–1378. [Google Scholar] [CrossRef]
- Jin, Z.; Tietjen, I.; Bu, L.; Liu-Yesucevitz, L.; Gaur, S.K.; Walsh, C.A.; Piao, X. Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum. Mol. Genet. 2007, 16, 1972–1985. [Google Scholar] [CrossRef]
- Shashidhar, S.; Lorente, G.; Nagavarapu, U.; Nelson, A.; Kuo, J.; Cummins, J.; Nikolich, K.; Urfer, R.; Foehr, E.D. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 2005, 24, 1673–1682. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Thomazy, V. Protein Crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect. Tissue Res. 2000, 41, 1–27. [Google Scholar] [CrossRef]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef]
- Lorand, L.; Iismaa, S.E. Transglutaminase diseases: From biochemistry to the bedside. FASEB J. 2019, 33, 3–12. [Google Scholar] [CrossRef]
- Boros, S.; Xi, Q.; Dimke, H.; van der Kemp, A.W.; Tudpor, K.; Verkaart, S.; Lee, K.P.; Bindels, R.J.; Hoenderop, J.G. Tissue transglutaminase inhibits the TRPV5-dependent calcium transport in an N-glycosylation-dependent manner. Cell. Mol. Life Sci. 2012, 69, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Janiak, A.; Zemskov, E.A.; Belkin, A.M. Cell Surface Transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src–p190RhoGAP signaling pathway. Mol. Biol. Cell 2006, 17, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Xu, J.; Sung, B.; Kumar, S.; Yu, D.; Aggarwal, B.B.; Mehta, K. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2012, 14, R4. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Friedland, S.; Corson, N.; Xu, L. GPR56 inhibits melanoma growth by internalizing and degrading its ligand TG2. Cancer Res. 2014, 74, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L. Transglutaminase 2 takes center stage as a cancer cell survival factor and therapy target. Mol. Carcinog. 2019, 58, 837–853. [Google Scholar] [CrossRef]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef]
- Arnaout, M.; Mahalingam, B.; Xiong, J.-P. INTEGRIN STRUCTURE, ALLOSTERY, AND BIDIRECTIONAL SIGNALING. Annu. Rev. Cell Dev. Biol. 2005, 21, 381–410. [Google Scholar] [CrossRef]
- Zemskov, E.A.; Loukinova, E.; Mikhailenko, I.; Coleman, R.A.; Strickland, D.K.; Belkin, A.M. Regulation of platelet-derived growth factor receptor function by integrin-associated cell surface transglutaminase. J. Biol. Chem. 2009, 284, 16693–16703. [Google Scholar] [CrossRef]
- Jeong, S.-J.; Luo, R.; Singer, K.; Giera, S.; Kreidberg, J.; Kiyozumi, D.; Shimono, C.; Sekiguchi, K.; Piao, X. GPR56 functions together with α3β1 integrin in regulating cerebral cortical development. PLoS ONE 2013, 8, e68781. [Google Scholar] [CrossRef]
- Kim, J.-E.; Han, J.M.; Park, C.R.; Shin, K.-J.; Ahn, C.; Seong, J.Y.; Hwang, J.-I. Splicing variants of the orphan G-protein-coupled receptor GPR56 regulate the activity of transcription factors associated with tumorigenesis. J. Cancer Res. Clin. Oncol. 2010, 136, 47–53. [Google Scholar] [CrossRef]
- Salzman, G.S.; Zhang, S.; Fernandez, C.G.; Araç, D.; Koide, S. Specific and direct modulation of the interaction between adhesion GPCR GPR56/ADGRG1 and tissue transglutaminase 2 using synthetic ligands. Sci. Rep. 2020, 10, 16912. [Google Scholar] [CrossRef]
- Inoue, A.; Ishiguro, J.; Kitamura, H.; Arima, N.; Okutani, M.; Shuto, A.; Higashiyama, S.; Ohwada, T.; Arai, H.; Makide, K.; et al. TGFα shedding assay: An accurate and versatile method for detecting GPCR activation. Nat. Methods 2012, 9, 1021–1029. [Google Scholar] [CrossRef]
- Kleinau, G.; Neumann, S.; Grüters, A.; Krude, H.; Biebermann, H. Novel insights on thyroid-stimulating hormone receptor signal transduction. Endocr. Rev. 2013, 34, 691–724. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.-Y.; Chang, G.-W.; Huang, Y.-S.; Peng, Y.-M.; Hsiao, C.-C.; Kuo, M.-L.; Lin, H.-H. Heparin interacts with the adhesion GPCR GPR56, reduces receptor shedding, and promotes cell adhesion and motility. J. Cell Sci. 2016, 129, 2156–2169. [Google Scholar] [CrossRef]
- Silva, J.-P.; Lelianova, V.; Hopkins, C.; Volynski, K.E.; Ushkaryov, Y. Functional cross-interaction of the fragments produced by the cleavage of distinct adhesion G-protein-coupled receptors. J. Biol. Chem. 2009, 284, 6495–6506. [Google Scholar] [CrossRef]
- Prömel, S.; Frickenhaus, M.; Hughes, S.; Mestek, L.; Staunton, D.; Woollard, A.; Vakonakis, I.; Schöneberg, T.; Schnabel, R.; Russ, A.P.; et al. The GPS motif is a molecular switch for bimodal activities of adhesion class G protein-coupled receptors. Cell Rep. 2012, 2, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, G.; Mohanty, S.; Scott, G.; Fazal, F.; Rahman, A.; Begum, S.; Hynes, R.O.; Xu, L. GPR56 Regulates VEGF production and angiogenesis during melanoma progression. Cancer Res. 2011, 71, 5558–5568. [Google Scholar] [CrossRef]
- Scarpellini, A.; Germack, R.; Lortat-Jacob, H.; Muramatsu, T.; Billett, E.; Johnson, T.; Verderio, E.A. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J. Biol. Chem. 2009, 284, 18411–18423. [Google Scholar] [CrossRef] [PubMed]
- Iversen, R.; Di Niro, R.; Stamnaes, J.; Lundin, K.E.A.; Wilson, P.C.; Sollid, L.M. Transglutaminase 2–specific autoantibodies in celiac disease target clustered, N-terminal epitopes not displayed on the surface of cells. J. Immunol. 2013, 190, 5981–5991. [Google Scholar] [CrossRef]
- Drake, M.T.; Shenoy, S.K.; Lefkowitz, R.J. Trafficking of G Protein–Coupled Receptors. Circ. Res. 2006, 99, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Olaniru, O.E.; Cheng, J.; Ast, J.; Arvaniti, A.; Atanes, P.; Huang, G.C.; King, A.J.; Jones, P.M.; Broichhagen, J.; Hodson, D.J.; et al. SNAP-tag-enabled super-resolution imaging reveals constitutive and agonist-dependent trafficking of GPR56 in pancreatic β-cells. Mol. Metab. 2021, 53, 101285. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Luo, R.; Jin, P.; Li, T.; Oak, H.C.; Giera, S.; Monk, K.R.; Lak, P.; Shoichet, B.K.; Piao, X. GAIN domain–mediated cleavage is required for activation of G protein–coupled receptor 56 (GPR56) by its natural ligands and a small-molecule agonist. J. Biol. Chem. 2019, 294, 19246–19254. [Google Scholar] [CrossRef] [PubMed]
- Yiu, T.W.; Holman, S.R.; Kaidonis, X.; Graham, R.M.; Iismaa, S.E. Transglutaminase 2 Facilitates Murine Wound Healing in a Strain-Dependent Manner. Int. J. Mol. Sci. 2023, 24, 11475. [Google Scholar] [CrossRef]
- Haroon, Z.A.; Hettasch, J.M.; Lai, T.-S.; Dewhirst, M.W.; Greenberg, C.S. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999, 13, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, M.; Höpfner, B.; Aeschlimann, D.; Lüthi, U.; Meuli, M.; Altermatt, S.; Gobet, R.; Bruckner-Tuderman, L.; Steinmann, B. Cross-linking of the dermo-epidermal junction of skin regenerating from keratinocyte autografts. Anchoring fibrils are a target for tissue transglutaminase. J. Clin. Investig. 1996, 98, 1174–1184. [Google Scholar] [CrossRef]
- Sud, N.; Sharma, R.; Ray, R.; Chattopadhyay, T.K.; Ralhan, R. Differential expression of G-protein coupled receptor 56 in human esophageal squamous cell carcinoma. Cancer Lett. 2006, 233, 265–270. [Google Scholar] [CrossRef] [PubMed]
- McSharry, B.P.; Jones, C.J.; Skinner, J.W.; Kipling, D.; Wilkinson, G.W.G. Human telomerase reverse transcriptase-immortalized MRC-5 and HCA2 human fibroblasts are fully permissive for human cytomegalovirus. J. Gen. Virol. 2001, 82, 855–863. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Aeschlimann, P.; Strigun, A.; Sanders, D.S.; Woodroofe, N.; Aeschlimann, D. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann. Neurol. 2008, 64, 332–343. [Google Scholar] [CrossRef]
- Thomas, H.; Beck, K.; Adamczyk, M.; Aeschlimann, P.; Langley, M.; Oita, R.C.; Thiebach, L.; Hils, M.; Aeschlimann, D. Transglutaminase 6: A protein associated with central nervous system development and motor function. Amino Acids 2013, 44, 161–177. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Koeller, M.K.; Allen-Hoffmann, B.L.; Mosher, D.F. Isolation of a cDNA encoding a novel member of the transglutaminase gene family from human keratinocytes. Detection and identification of transglutaminase gene products based on reverse transcription-polymerase chain reaction with degenerate primers. J. Biol. Chem. 1998, 273, 3452–3460. [Google Scholar] [CrossRef]
- Ali, N.; Knaüper, V. Phorbol Ester-induced shedding of the prostate cancer marker transmembrane protein with epidermal growth factor and two follistatin motifs 2 is mediated by the disintegrin and metalloproteinase-17. J. Biol. Chem. 2007, 282, 37378–37388. [Google Scholar] [CrossRef] [PubMed]
- Knäuper, V.; Patterson, M.L.; Gomis-Rüth, F.X.; Smith, B.; Lyons, A.; Docherty, A.J.; Murphy, G. The role of exon 5 in fibroblast collagenase (MMP-1) substrate specificity and inhibitor selectivity. Eur. J. Biochem. 2001, 268, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Kielty, C.M.; Phillips, J.E.; Child, A.H.; Pope, F.; Shuttleworth, C. Fibrillin secretion and microfibril assembly by Marfan dermal fibroblasts. Matrix Biol. 1994, 14, 191–199. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, L.; Edwards, J.; Heil, A.; Dewitt, S.; Biebermann, H.; Aeschlimann, D.; Knäuper, V. Mesenchymal Transglutaminase 2 Activates Epithelial ADAM17: Link to G-Protein-Coupled Receptor 56 (ADGRG1) Signalling. Int. J. Mol. Sci. 2024, 25, 2329. https://doi.org/10.3390/ijms25042329
Bauer L, Edwards J, Heil A, Dewitt S, Biebermann H, Aeschlimann D, Knäuper V. Mesenchymal Transglutaminase 2 Activates Epithelial ADAM17: Link to G-Protein-Coupled Receptor 56 (ADGRG1) Signalling. International Journal of Molecular Sciences. 2024; 25(4):2329. https://doi.org/10.3390/ijms25042329
Chicago/Turabian StyleBauer, Lea, Jessica Edwards, Andreas Heil, Sharon Dewitt, Heike Biebermann, Daniel Aeschlimann, and Vera Knäuper. 2024. "Mesenchymal Transglutaminase 2 Activates Epithelial ADAM17: Link to G-Protein-Coupled Receptor 56 (ADGRG1) Signalling" International Journal of Molecular Sciences 25, no. 4: 2329. https://doi.org/10.3390/ijms25042329
APA StyleBauer, L., Edwards, J., Heil, A., Dewitt, S., Biebermann, H., Aeschlimann, D., & Knäuper, V. (2024). Mesenchymal Transglutaminase 2 Activates Epithelial ADAM17: Link to G-Protein-Coupled Receptor 56 (ADGRG1) Signalling. International Journal of Molecular Sciences, 25(4), 2329. https://doi.org/10.3390/ijms25042329