

Multiple Lines of Evidence Support 199 SARS-CoV-2 Positively Selected Amino Acid Sites

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
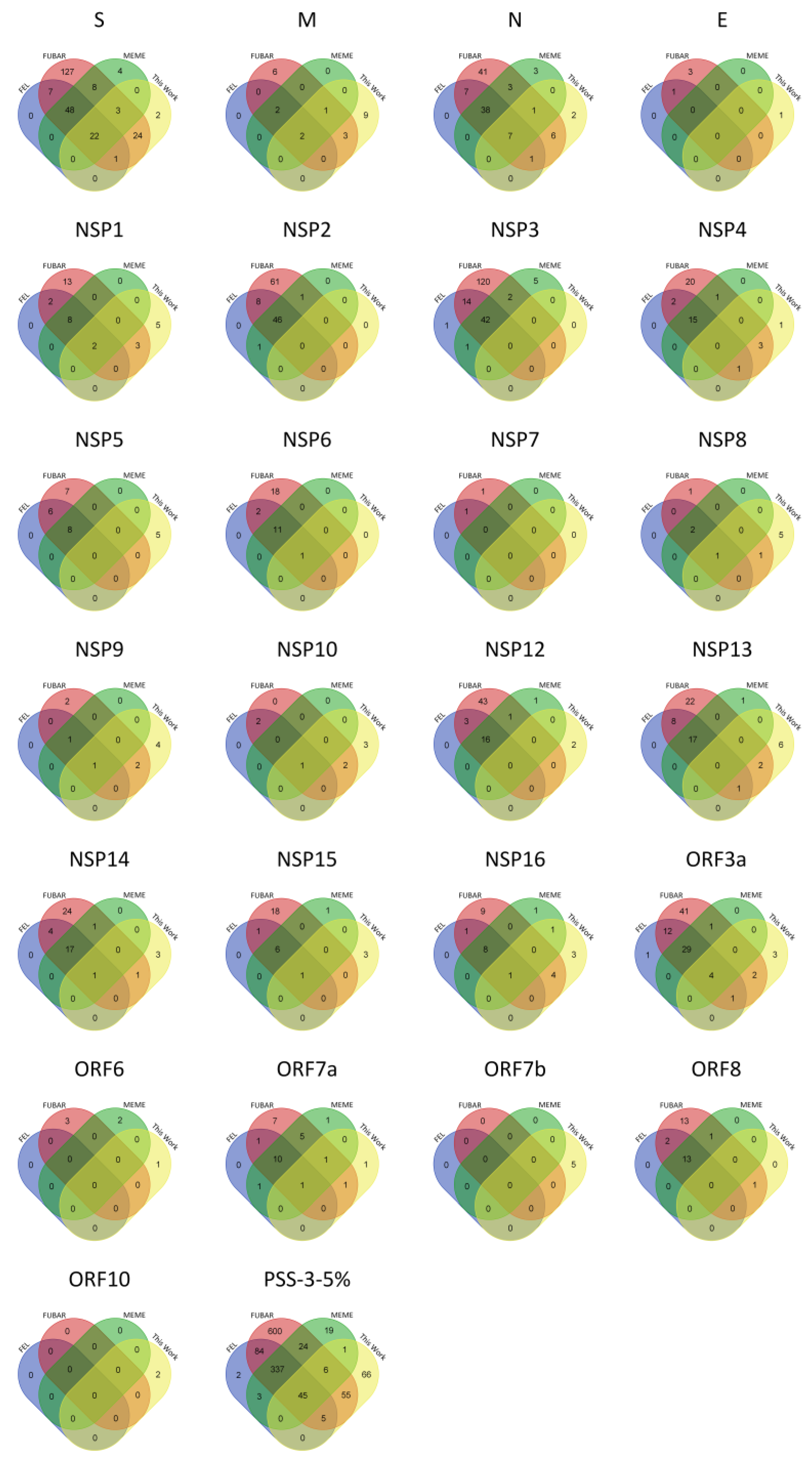
2.1. PSS Inferences in SARS-CoV-2
2.2. PSS Inferences in Non-SARS-CoV-2 Coronaviruses
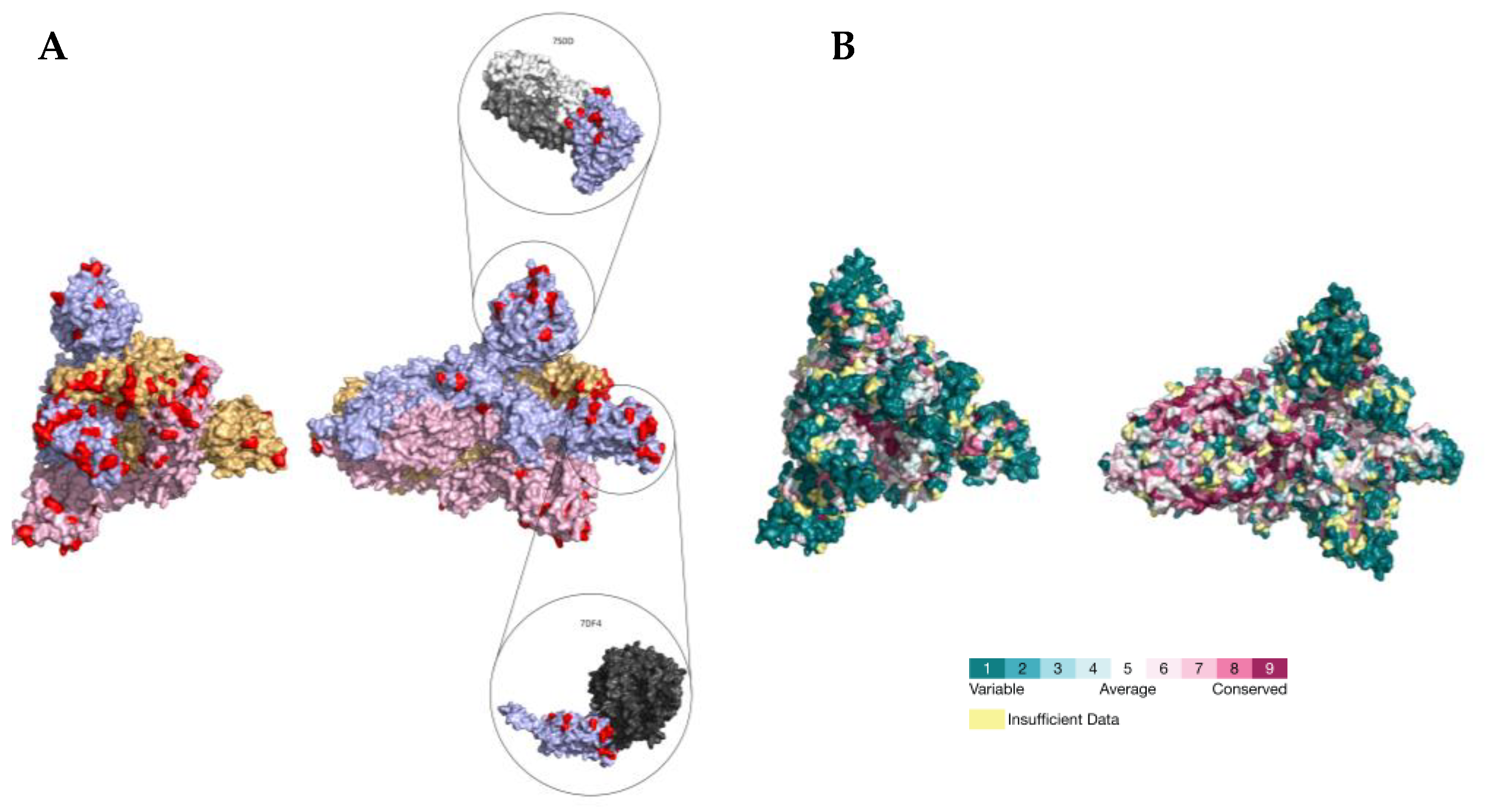
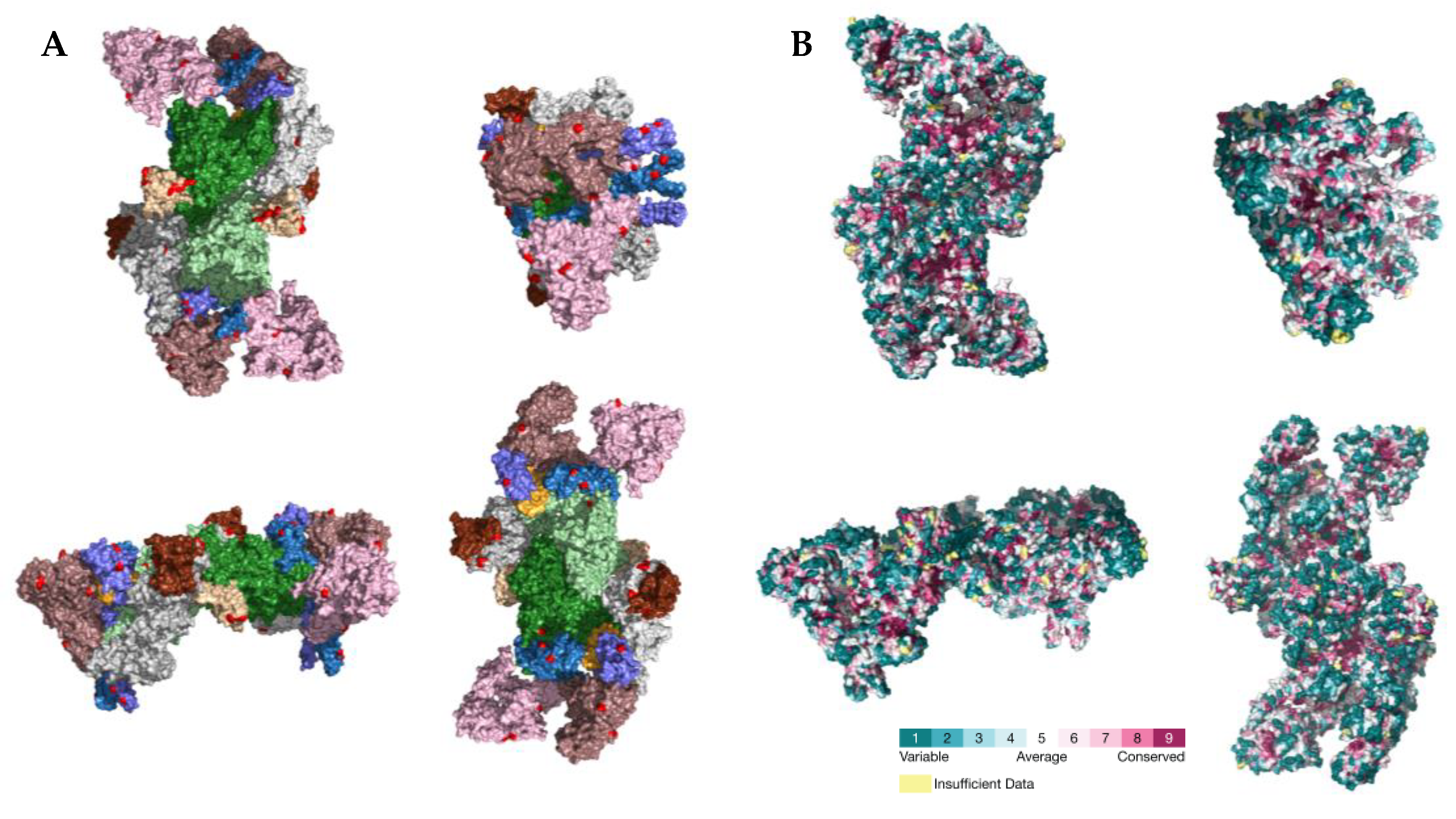
2.3. PSS Location on Protein Structures
3. Discussion
4. Materials and Methods
4.1. The Sequence Data for SARS-CoV-2
4.2. Sequence Data for Non-SARS-CoV-2 Coronavirus
4.3. PSS Detection
4.4. Orthologous Gene Identification
4.5. Mapping of PSS on Protein Structure Prediction
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tyrrell, D.A.; Bynoe, M.L. Cultivation of a novel type of common-cold virus in organ cultures. Br. Med. J. 1965, 1, 1467–1470. [Google Scholar] [CrossRef]
- Chan, J.F.; To, K.K.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Mesel-Lemoine, M.; Millet, J.; Vidalain, P.-O.; Law, H.; Vabret, A.; Lorin, V.; Escriou, N.; Albert, M.L.; Nal, B.; Tangy, F. A Human Coronavirus Responsible for the Common Cold Massively Kills Dendritic Cells but Not Monocytes. J. Virol. 2012, 86, 7577–7587. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Khan, M.; Haroon; Musa, T.H.; Nasir, S.; Hui, J.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. COVID-19: Zoonotic aspects. Travel Med. Infect. Dis. 2020, 36, 101607. [Google Scholar] [CrossRef] [PubMed]
- de Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses Academic Press; King, A., Adams, M., Cartens, E., Lefkowitz, E., Eds.; Elsevier Inc.: New York, NY, USA, 2012; pp. 806–828. [Google Scholar]
- Tseng, Y.T.; Chang, C.H.; Wang, S.M.; Huang, K.J.; Wang, C.T. Identifying SARS-CoV membrane protein amino acid residues linked to virus-like particle assembly. PLoS ONE 2013, 8, e64013. [Google Scholar] [CrossRef] [PubMed]
- Mahtarin, R.; Islam, S.; Islam, M.J.; Ullah, M.O.; Ali, M.A.; Halim, M.A. Structure and dynamics of membrane protein in SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 4725–4738. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wen, J.; Tang, L.; Zhang, H.; Zhang, X.; Li, Y.; Wang, J.; Han, Y.; Li, G.; Shi, J.; et al. The M protein of SARS-CoV: Basic structural and immunological properties. Genom. Proteom. Bioinform. 2003, 1, 118–130. [Google Scholar] [CrossRef]
- Voss, D.; Pfefferle, S.; Drosten, C.; Stevermann, L.; Traggiai, E.; Lanzavecchia, A.; Becker, S. Studies on membrane topology, N-glycosylation and functionality of SARS-CoV membrane protein. Virol. J. 2009, 6, 79. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Kubo, H.; Yamada, Y.K.; Taguchi, F. Localization of neutralizing epitopes and the receptor-binding site within the amino-terminal 330 amino acids of the murine coronavirus spike protein. J. Virol. 1994, 68, 5403–5410. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zeng, R.; von Brunn, A.; Lei, J. Structural characterization of the C-terminal domain of SARS-CoV-2 nucleocapsid protein. Mol. Biomed. 2020, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Chang, C.K.; Ikeya, T.; Güntert, P.; Chang, Y.H.; Hsu, Y.L.; Huang, T.H.; Kainosho, M. Solution structure of the c-terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL-NMR method. J. Mol. Biol. 2008, 380, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Is There a Link Between the Pathogenic Human Coronavirus Envelope Protein and Immunopathology? A Review of the Literature. Front. Microbiol. 2020, 11, 2086. [Google Scholar] [CrossRef] [PubMed]
- Zmasek, C.M.; Lefkowitz, E.J.; Niewiadomska, A.; Scheuermann, R.H. Genomic evolution of the Coronaviridae family. Virology 2022, 570, 123–133. [Google Scholar] [CrossRef]
- Fitch, W.M. Homology: A personal view on some of the problems. Trends Genet. 2000, 16, 227–231. [Google Scholar] [CrossRef]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573.e1513. [Google Scholar] [CrossRef]
- Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Nieto-Torres, J.L.; DeDiego, M.L.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Perlman, S.; Enjuanes, L. Identification of the Mechanisms Causing Reversion to Virulence in an Attenuated SARS-CoV for the Design of a Genetically Stable Vaccine. PLoS Pathog. 2015, 11, e1005215. [Google Scholar] [CrossRef]
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R., 3rd; Kuhn, P.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 2009, 83, 10314–10318. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef]
- Graham, R.L.; Sparks, J.S.; Eckerle, L.D.; Sims, A.C.; Denison, M.R. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008, 133, 88–100. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Ulasli, M.; Vonk, A.M.; Reggiori, F.; Rottier, P.J.; de Haan, C.A. Mobility and interactions of coronavirus nonstructural protein 4. J. Virol. 2011, 85, 4572–4577. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Sun, F.; Li, X.; Pang, H.; Xu, X.; Bartlam, M.; Rao, Z. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nat. Struct. Mol. Biol. 2005, 12, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Krishna, T.S.; Kong, X.P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Biswas, N.; Kumar, K.; Mallick, P.; Das, S.; Kamal, I.M.; Bose, S.; Choudhury, A.; Chakrabarti, S. Structural and Drug Screening Analysis of the Non-structural Proteins of Severe Acute Respiratory Syndrome Coronavirus 2 Virus Extracted From Indian Coronavirus Disease 2019 Patients. Front. Genet. 2021, 12, 626642. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, J.; Sun, Y.; Wu, A.; Su, C.; Gao, G.; Cai, H.; Qiu, S.; Wu, Y.; Ahola, T.; et al. Structure-function analysis of severe acute respiratory syndrome coronavirus RNA cap guanine-N7-methyltransferase. J. Virol. 2013, 87, 6296–6305. [Google Scholar] [CrossRef]
- Lin, S.; Chen, H.; Ye, F.; Chen, Z.; Yang, F.; Zheng, Y.; Cao, Y.; Qiao, J.; Yang, S.; Lu, G. Crystal structure of SARS-CoV-2 nsp10/nsp16 2′-O-methylase and its implication on antiviral drug design. Signal. Transduct. Target Ther. 2020, 5, 131. [Google Scholar] [CrossRef]
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gao, M.; Gao, X.; Zhu, B.; Huang, J.; Luo, K.; Zhang, Y.; Sun, J.; Deng, M.; Lou, Z. SARS-CoV-2 non-structural protein 13 (nsp13) hijacks host deubiquitinase USP13 and counteracts host antiviral immune response. Signal. Transduct. Target Ther. 2021, 6, 119. [Google Scholar] [CrossRef]
- Pillon, M.C.; Frazier, M.N.; Dillard, L.B.; Williams, J.G.; Kocaman, S.; Krahn, J.M.; Perera, L.; Hayne, C.K.; Gordon, J.; Stewart, Z.D.; et al. Cryo-EM structures of the SARS-CoV-2 endoribonuclease Nsp15 reveal insight into nuclease specificity and dynamics. Nat. Commun. 2021, 12, 636. [Google Scholar] [CrossRef]
- Ricagno, S.; Coutard, B.; Grisel, S.; Brémond, N.; Dalle, K.; Tocque, F.; Campanacci, V.; Lichière, J.; Lantez, V.; Debarnot, C.; et al. Crystallization and preliminary X-ray diffraction analysis of Nsp15 from SARS coronavirus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 409–411. [Google Scholar] [CrossRef]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and Function of Major SARS-CoV-2 and SARS-CoV Proteins. Bioinform. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Soares, R.; Vieira, C.P.; Vieira, J. Predictive Models of within- and between-Species SARS-CoV-2 Transmissibility. Viruses 2022, 14, 1565. [Google Scholar] [CrossRef]
- Nielsen, R. Statistical tests of selective neutrality in the age of genomics. Heredity 2001, 86, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pöhlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef]
- Carvalho, P.P.D.; Alves, N. Featuring ACE2 Binding SARS-CoV and SARS-CoV-2 through a Conserved Evolutionary Pattern of Amino Acid Residues. J. Biomol. Struct. Dyn. 2022, 40, 11719–11728. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Liu, Y.; Zhou, Z.; Zhang, Z.; Xiao, X.; Liu, Z.; Chen, A.; Dong, X.; Tian, F.; Chen, S.; et al. Genome-wide CRISPR activation screen identifies candidate receptors for SARS-CoV-2 entry. Sci. China Life Sci. 2022, 65, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html?fbclid=IwAR3YwLYIOMz431yqTo-ZyEiG8V0ruYv_XkrgOZzZUzC77nbROg11bUfm5Qg (accessed on 6 May 2021).
- Holmes, E.C. The Evolutionary Genetics of Emerging Viruses. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 353–372. [Google Scholar] [CrossRef]
- Miras, M.; Sempere, R.N.; Kraft, J.J.; Miller, W.A.; Aranda, M.A.; Truniger, V. Interfamilial recombination between viruses led to acquisition of a novel translation-enhancing RNA element that allows resistance breaking. New Phytol. 2014, 202, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further Evidence for Bats as the Evolutionary Source of Middle East Respiratory Syndrome Coronavirus. mBio 2017, 8, e00373-17. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [PubMed]
- Gire, S.K.; Goba, A.; Andersen, K.G.; Sealfon, R.S.; Park, D.J.; Kanneh, L.; Jalloh, S.; Momoh, M.; Fullah, M.; Dudas, G.; et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014, 345, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Li, Y. Global evolutionary history and spatio-temporal dynamics of dengue virus type 2. Sci. Rep. 2017, 7, 45505. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Front. Microbiol. 2020, 11, 550674. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Feng, Y.; Yi, J.; Yang, L.; Wang, Y.; Wen, J.; Zhao, W.; Kim, P.; Zhou, X. COV2Var, a function annotation database of SARS-CoV-2 genetic variation. Nucleic Acids Res. 2023, 52, gkad958. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Rahimian, K.; Arefian, E.; Mahdavi, B.; Mahmanzar, M.; Kuehu, D.L.; Deng, Y. SARS2Mutant: SARS-CoV-2 amino-acid mutation atlas database. NAR Genom. Bioinform. 2023, 5, lqad037. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Qian, Z.; Lu, X.; Lu, J. Adaptive Evolution of the Spike Protein in Coronaviruses. Mol. Biol. Evol. 2023, 40, msad089. [Google Scholar] [CrossRef] [PubMed]
- Ben Chorin, A.; Masrati, G.; Kessel, A.; Narunsky, A.; Sprinzak, J.; Lahav, S.; Ashkenazy, H.; Ben-Tal, N. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. 2020, 29, 258–267. [Google Scholar] [CrossRef]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef]
- Wintachai, P.; Wikan, N.; Kuadkitkan, A.; Jaimipuk, T.; Ubol, S.; Pulmanausahakul, R.; Auewarakul, P.; Kasinrerk, W.; Weng, W.-Y.; Panyasrivanit, M.; et al. Identification of prohibitin as a Chikungunya virus receptor protein. J. Med. Virol. 2012, 84, 1757–1770. [Google Scholar] [CrossRef]
- Kuadkitkan, A.; Wikan, N.; Fongsaran, C.; Smith, D.R. Identification and characterization of prohibitin as a receptor protein mediating DENV-2 entry into insect cells. Virology 2010, 406, 149–161. [Google Scholar] [CrossRef]
- Lopez-Fernandez, H.; Duque, P.; Vazquez, N.; Fdez-Riverola, F.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. SEDA: A Desktop Tool Suite for FASTA Files Processing. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 1850–1860. [Google Scholar] [CrossRef]
- López-Fernández, H.; Ferreira, P.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. The pegi3s Bioinformatics Docker Images Project. In Proceedings of the Practical Applications of Computational Biology & Bioinformatics, 15th International Conference (PACBB 2021), Salamanca, Spain, 6–8 October 2021; pp. 31–40. [Google Scholar]
- López-Fernández, H.; Vieira, C.P.; Ferreira, P.; Gouveia, P.; Fdez-Riverola, F.; Reboiro-Jato, M.; Vieira, J. On the Identification of Clinically Relevant Bacterial Amino Acid Changes at the Whole Genome Level Using Auto-PSS-Genome. Interdiscip. Sci. Comput. Life Sci. 2021, 13, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. Available online: http://www.pymol.org/pymol (accessed on 1 January 2023).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Length | VAAS | PSS-FEL | PSS-FUBAR | PSS-MEME | PSS-3 | #PSS-3-5% |
|---|---|---|---|---|---|---|---|
| S | 1273 | 481 (37.8) | 181 (37.6) | 327 (68.0) | 352 (73.2) | 173 (36.0) | 70 (14.6) |
| E | 75 | 21 (28.0) | 1 (4.8) | 12 (57.1) | 4 (19.0) | 1 (4.8) | 0 (0.0) |
| M | 222 | 59 (26.6) | 14 (23.7) | 30 (50.8) | 29 (49.2) | 12 (20.3) | 4 (6.8) |
| N | 419 | 231 (55.1) | 109 (47.2) | 166 (71.9) | 179 (77.5) | 104 (45.0) | 45 (19.5) |
| NSP1 | 180 | 97 (53.9) | 43 (44.3) | 69 (71.1) | 65 (67.0) | 33 (34.0) | 10 (10.3) |
| NSP2 | 638 | 341 (53.4) | 92 (27.0) | 183 (53.7) | 197 (57.8) | 86 (25.2) | 46 (13.5) |
| NSP3 | 1945 | 801 (41.2) | 138 (17.2) | 325 (40.6) | 398 (49.7) | 118 (14.7) | 42 (5.2) |
| NSP4 | 500 | 159 (31.8) | 31 (19.5) | 74 (46.5) | 83 (52.2) | 30 (18.9) | 15 (9.4) |
| NSP5 | 306 | 83 (27.1) | 19 (22.9) | 37 (44.6) | 42 (50.6) | 16 (19.3) | 8 (9.6) |
| NSP6 | 290 | 120 (41.4) | 24 (20.0) | 58 (48.3) | 56 (46.7) | 24 (20.0) | 12 (10.0) |
| NSP7 | 83 | 28 (33.7) | 4 (14.3) | 9 (32.1) | 9 (32.1) | 3 (10.7) | 0 (0.0) |
| NSP8 | 198 | 60 (30.3) | 7 (11.7) | 22 (36.7) | 13 (21.7) | 5 (8.3) | 3 (5.0) |
| NSP9 | 113 | 36 (31.9) | 8 (22.2) | 16 (44.4) | 8 (22.2) | 5 (13.9) | 2 (5.6) |
| NSP10 | 139 | 33 (23.7) | 3 (9.1) | 13 (39.4) | 7 (21.2) | 2 (6.1) | 1 (3.0) |
| NSP12 | 932 | 256 (27.5) | 34 (13.3) | 100 (39.1) | 94 (36.7) | 30 (11.7) | 16 (6.3) |
| NSP13 | 601 | 186 (30.9) | 42 (22.6) | 87 (46.8) | 81 (43.5) | 36 (19.4) | 17 (9.1) |
| NSP14 | 527 | 181 (34.3) | 46 (25.4) | 91 (50.3) | 85 (47.0) | 42 (23.2) | 18 (9.9) |
| NSP15 | 346 | 125 (36.1) | 17 (13.6) | 52 (41.6) | 55 (44.0) | 14 (11.2) | 7 (5.6) |
| NSP16 | 298 | 90 (30.2) | 15 (16.7) | 39 (43.3) | 58 (64.4) | 15 (16.7) | 9 (10.0) |
| Orf3a | 275 | 183 (66.5) | 89 (48.6) | 135 (73.8) | 144 (78.7) | 86 (47.0) | 33 (12.0) |
| Orf6 | 61 | 31 (50.8) | 7 (22.6) | 14 (45.2) | 22 (71.0) | 6 (19.4) | 0 (0.0) |
| Orf7a | 121 | 86 (71.1) | 29 (33.7) | 51 (59.3) | 82 (95.3) | 29 (33.7) | 10 (8.3) |
| Orf7b | 43 | 28 (65.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Orf8 | 121 | 87 (71.9) | 35 (40.2) | 53 (60.9) | 72 (82.8) | 33 (37.9) | 13 (10.7) |
| Orf10 | 38 | 24 (63.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| TOTAL | 3827 | 988 (25.8) | 1963 (51.3) | 2135 (55.8) | 903 (23.6) | 381 (10.0) |
| Protein | Datasets | SARS-CoV-2 Positions | |
|---|---|---|---|
| S | PEDV–Betacoronavirus1 | 75 | |
| PEDV–Alphacoronavirus1 | Gap 97-98 | ||
| M | Unknown Bat-CoV–Bat-CoV-HKU9– Bat-CoV-HKU10 | 4 | |
| Bat-CoV-HKU2–Bat-CoV-HKU9– Bat-CoV-HKU10–Alphacoronavirus1 | 3 | ||
| Bat-CoV-HKU2–Bat-CoV-HKU10 | 6 | ||
| N | Porcine-CoV-HKU15–Bat-CoV-HKU9 | 62 | |
| Alphacoronavirus1–hCoV-HKU1 | 91 | ||
| hCoV-HKU1–Murine-CoV | 289 | ||
| ORF1ab | NSP3 | hCoV-HKU1–hCoV-NL63 | 112 |
| Murine-CoV–PEDV | 162 | ||
| Murine-CoV–Betacoronavirus1 | 1234 | ||
| NSP6 | Murine-CoV–PEDV | 138 | |
| NSP12 | MERS-CoV–PEDV | 9 | |
| Protein | PSS-FUBAR | PSS-codeML | PSS Common | |
|---|---|---|---|---|
| Structural | S | 50 (35) | 51 (37) | 15 (13) |
| M | 12 (8) | 13 (7) | 4 (4) | |
| N | 25 (18) | 11 (9) | 4 (4) | |
| E | 1 (1) | NA | NA | |
| Non-Structural | nsp1 | 8 (4) | 4 (1) | 2 (1) |
| nsp2 | 15 (14) | 2 (2) | NA | |
| nsp3 | 58 (52) | 8 (6) | 4 (3) | |
| nsp4 | 5 (5) | 2 (2) | 1 (1) | |
| nsp5 | 3 (3) | NA | NA | |
| nsp6 | 7 (7) | NA | NA | |
| nsp7 | 1 (1) | NA | NA | |
| nsp8 | 3 (3) | NA | NA | |
| nsp9 | NA | NA | NA | |
| nsp10 | 1 (1) | NA | NA | |
| nsp12 | 9 (6) | 2 (2) | 1 (1) | |
| nsp13 | 1 (1) | 1 (1) | NA | |
| nsp14 | 2 (2) | NA | NA | |
| nsp15 | 9 (8) | 2 (2) | 2 (2) | |
| nsp16 | 6 (5) | NA | NA | |
| Accessory | 44 | 5 | 2 | |
| TOTAL | 260 (174) | 101 (69) | 35 (29) | |
| # of Sites (%) | |
|---|---|
| GISAID list | 123 |
| Located in RBD | 34 (27.6) |
| Located in NTD | 54 (43.9) |
| SARS-CoV-2 PSSs | 52 |
| GISAID sites | 52 (100) |
| Predicted by CoVs PSSs | 22 (42.3) |
| Located in RBD | 22 |
| Predicted by CoVs PSSs | 9 (40,9) |
| Located in NTD | 17 |
| Predicted by CoVs PSSs | 10 (58.8) |
| Datasets | Genus | Size (Number of Input Files) |
|---|---|---|
| Bat-CoV-HKU10 | αCoV | 8 |
| Rhinolophus-Bat-COV-HKU2 | αCoV | 8 |
| Pipistrellus-Bat-CoV-HKU5 | βCoV | 9 |
| Rousettus-Bat-CoV-HKU9 | βCoV | 10 |
| Tylonycteris-Bat-CoV-HKU4 | βCoV | 10 |
| Unknown-Bat-CoV | αCoV | 19 |
| Murine-CoV | βCoV | 29 |
| Human-CoV-HKU1 | βCoV | 38 |
| Human-CoV-229E | αCoV | 44 |
| Human-CoV-NL63 | αCoV | 58 |
| Porcine-CoV-HKU15 | δCoV | 60 |
| Alphacoronavirus-1 | αCoV | 118 |
| MERS-CoV | βCoV | 219 |
| Betacoronaviruses-1 | βCoV | 310 |
| Porcine-epidemic-diarrhea-virus (PEDV) | αCoV | 690 |
| α-coronaviruses | 945 | |
| β-coronaviruses | 625 | |
| δ-coronaviruses | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, P.; Soares, R.; López-Fernández, H.; Vazquez, N.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. Multiple Lines of Evidence Support 199 SARS-CoV-2 Positively Selected Amino Acid Sites. Int. J. Mol. Sci. 2024, 25, 2428. https://doi.org/10.3390/ijms25042428
Ferreira P, Soares R, López-Fernández H, Vazquez N, Reboiro-Jato M, Vieira CP, Vieira J. Multiple Lines of Evidence Support 199 SARS-CoV-2 Positively Selected Amino Acid Sites. International Journal of Molecular Sciences. 2024; 25(4):2428. https://doi.org/10.3390/ijms25042428
Chicago/Turabian StyleFerreira, Pedro, Ricardo Soares, Hugo López-Fernández, Noé Vazquez, Miguel Reboiro-Jato, Cristina P. Vieira, and Jorge Vieira. 2024. "Multiple Lines of Evidence Support 199 SARS-CoV-2 Positively Selected Amino Acid Sites" International Journal of Molecular Sciences 25, no. 4: 2428. https://doi.org/10.3390/ijms25042428
APA StyleFerreira, P., Soares, R., López-Fernández, H., Vazquez, N., Reboiro-Jato, M., Vieira, C. P., & Vieira, J. (2024). Multiple Lines of Evidence Support 199 SARS-CoV-2 Positively Selected Amino Acid Sites. International Journal of Molecular Sciences, 25(4), 2428. https://doi.org/10.3390/ijms25042428