
Deep Flow Cytometry Unveils Distinct Immune Cell Subsets in Inducible T Cell Co-Stimulator Ligand (ICOSL)- and ICOS-Knockout Mice during Experimental Autoimmune Encephalomyelitis

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. ICOSL Deficiency Exacerbates EAE and Impacts Recovery during Remission
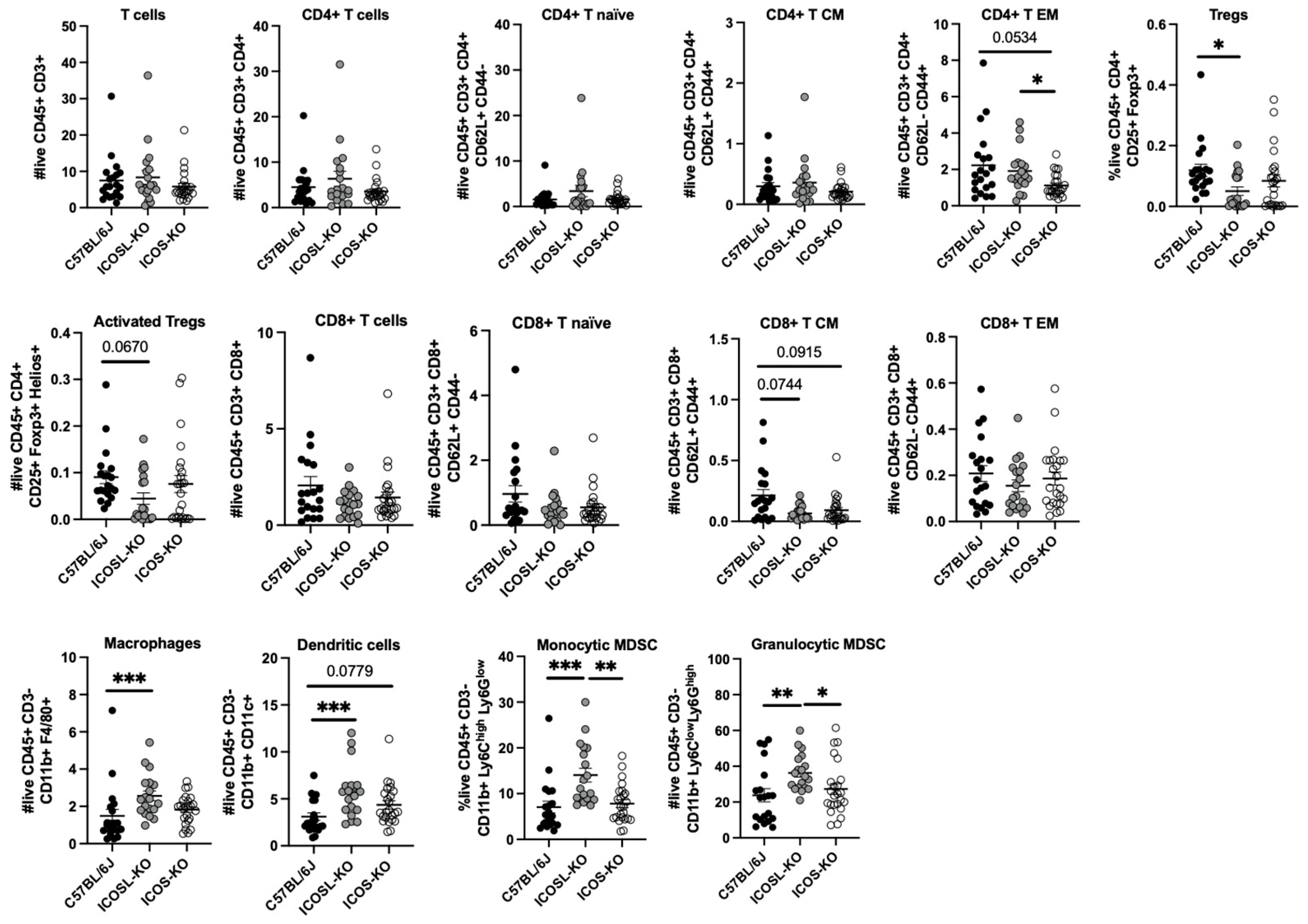
2.2. Deep Multiparametric Flow Cytometry Analysis with Classical Gating Strategy Reveals a Reduction in CD8+ TCM Cells and Tregs and an Increase in CD4+ TEM and Myeloid Cells in ICOSL-KO Mice
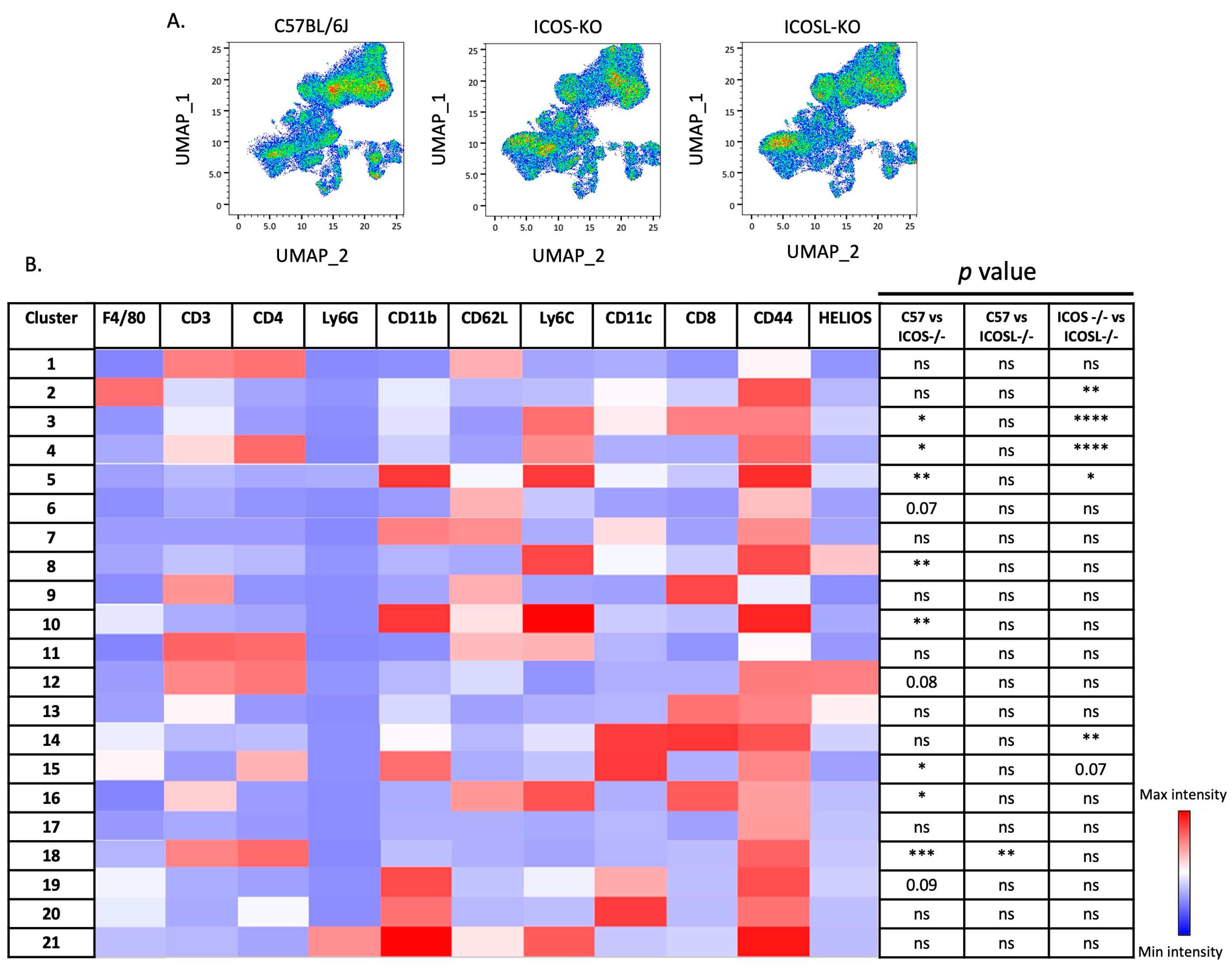
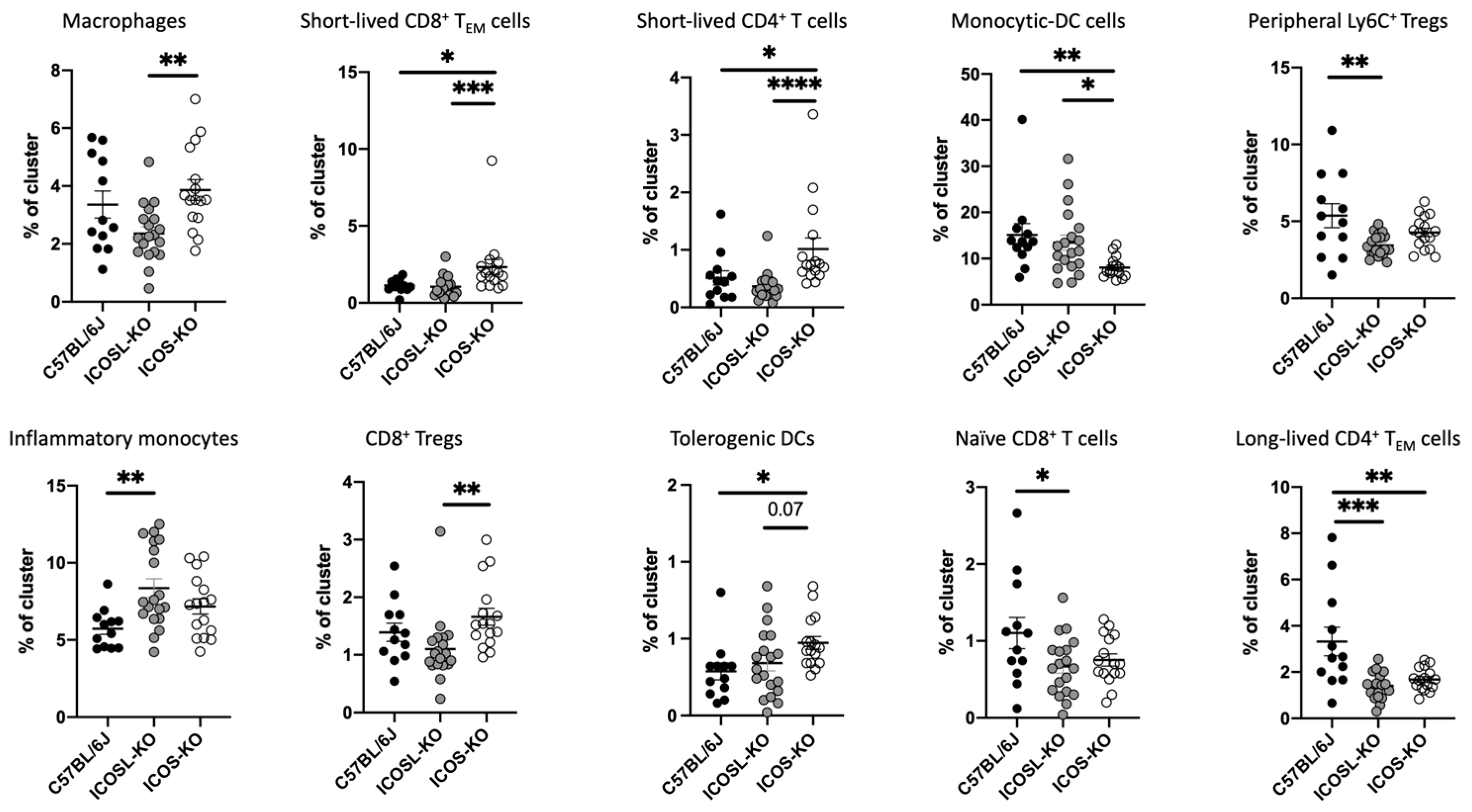
2.3. Deep Flow Cytometry Unsupervised Analysis Reveals Distinct Cell Subsets and New Clusters Differentially Associated with ICOSL-Driven Disease
3. Discussion
4. Materials and Methods
4.1. Induction and Clinical Evaluation of Experimental Autoimmune Encephalomyelitis (EAE)
4.2. MOG35–55-Induced Cytokine Release
4.3. Analysis of IL-17 Expression in Brain and Spinal Cord Tissues
4.4. Flow Cytometry
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, C.; Juedes, A.E.; Temann, U.A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef]
- Hedl, M.; Lahiri, A.; Ning, K.; Cho, J.H.; Abraham, C. Pattern recognition receptor signaling in human dendritic cells is enhanced by ICOS ligand and modulated by the Crohn’s disease ICOSLG risk allele. Immunity 2014, 40, 734–746. [Google Scholar] [CrossRef]
- Swallow, M.M.; Wallin, J.J.; Sha, W.C. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity 1999, 11, 423–432. [Google Scholar] [CrossRef]
- Galicia, G.; Kasran, A.; Uyttenhove, C.; De Swert, K.; Van Snick, J.; Ceuppens, J.L. ICOS deficiency results in exacerbated IL-17 mediated experimental autoimmune encephalomyelitis. J. Clin. Immunol. 2009, 29, 426–433. [Google Scholar] [CrossRef]
- Rottman, J.B.; Smith, T.; Tonra, J.R.; Ganley, K.; Bloom, T.; Silva, R.; Pierce, B.; Gutierrez-Ramos, J.C.; Ozkaynak, E.; Coyle, A.J. The costimulatory molecule ICOS plays an important role in the immunopathogenesis of EAE. Nat. Immunol. 2001, 2, 605–611. [Google Scholar] [CrossRef]
- Sporici, R.A.; Beswick, R.L.; von Allmen, C.; Rumbley, C.A.; Hayden-Ledbetter, M.; Ledbetter, J.A.; Perrin, P.J. ICOS ligand costimulation is required for T-cell encephalitogenicity. Clin. Immunol. 2001, 100, 277–288. [Google Scholar] [CrossRef]
- Dianzani, C.; Minelli, R.; Gigliotti, C.L.; Occhipinti, S.; Giovarelli, M.; Conti, L.; Boggio, E.; Shivakumar, Y.; Baldanzi, G.; Malacarne, V.; et al. B7h triggering inhibits the migration of tumor cell lines. J. Immunol. 2014, 192, 4921–4931. [Google Scholar] [CrossRef]
- Dianzani, C.; Minelli, R.; Mesturini, R.; Chiocchetti, A.; Barrera, G.; Boscolo, S.; Sarasso, C.; Gigliotti, C.L.; Sblattero, D.; Yagi, J.; et al. B7h triggering inhibits umbilical vascular endothelial cell adhesiveness to tumor cell lines and polymorphonuclear cells. J. Immunol. 2010, 185, 3970–3979. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, C.L.; Boggio, E.; Clemente, N.; Shivakumar, Y.; Toth, E.; Sblattero, D.; D’Amelio, P.; Isaia, G.C.; Dianzani, C.; Yagi, J.; et al. ICOS-Ligand Triggering Impairs Osteoclast Differentiation and Function In Vitro and In Vivo. J. Immunol. 2016, 197, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, S.; Dianzani, C.; Chiocchetti, A.; Boggio, E.; Clemente, N.; Gigliotti, C.L.; Soluri, M.F.; Minelli, R.; Fantozzi, R.; Yagi, J.; et al. Triggering of B7h by the ICOS modulates maturation and migration of monocyte-derived dendritic cells. J. Immunol. 2013, 190, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Raineri, D.; Dianzani, C.; Cappellano, G.; Maione, F.; Baldanzi, G.; Iacobucci, I.; Clemente, N.; Baldone, G.; Boggio, E.; Gigliotti, C.L.; et al. Osteopontin binds ICOSL promoting tumor metastasis. Commun. Biol. 2020, 3, 615. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.H.; Cao, Y.; Mangos, S.; Tardi, N.J.; Dande, R.R.; Lee, H.W.; Samelko, B.; Altintas, M.M.; Schmitz, V.P.; Lee, H.; et al. Nonimmune cell-derived ICOS ligand functions as a renoprotective αvβ3 integrin-selective antagonist. J. Clin. Invest. 2019, 129, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Raineri, D.; Cappellano, G.; Vilardo, B.; Maione, F.; Clemente, N.; Canciani, E.; Boggio, E.; Gigliotti, C.L.; Monge, C.; Dianzani, C.; et al. Inducible T-Cell Costimulator Ligand Plays a Dual Role in Melanoma Metastasis upon Binding to Osteopontin or Inducible T-Cell Costimulator. Biomedicines 2021, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Horton, M.A. The alpha v beta 3 integrin “vitronectin receptor”. Int. J. Biochem. Cell Biol. 1997, 29, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Oksenberg, J.R. The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron 2006, 52, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Suchanek, G.; Ozawa, K. Histopathology and the blood-cerebrospinal fluid barrier in multiple sclerosis. Ann. Neurol. 1994, 36, S42–S46. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175. [Google Scholar] [CrossRef]
- Mesturini, R.; Gigliotti, C.L.; Orilieri, E.; Cappellano, G.; Soluri, M.F.; Boggio, E.; Woldetsadik, A.; Dianzani, C.; Sblattero, D.; Chiocchetti, A.; et al. Differential induction of IL-17, IL-10, and IL-9 in human T helper cells by B7h and B7.1. Cytokine 2013, 64, 322–330. [Google Scholar] [CrossRef]
- Mesturini, R.; Nicola, S.; Chiocchetti, A.; Bernardone, I.S.; Castelli, L.; Bensi, T.; Ferretti, M.; Comi, C.; Dong, C.; Rojo, J.M.; et al. ICOS cooperates with CD28, IL-2, and IFN-gamma and modulates activation of human naïve CD4+ T cells. Eur. J. Immunol. 2006, 36, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Yong, P.F.; Salzer, U.; Grimbacher, B. The role of costimulation in antibody deficiencies: ICOS and common variable immunodeficiency. Immunol. Rev. 2009, 229, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I. Regulation of immune and autoimmune responses by ICOS-B7h interaction. Clin. Immunol. 2005, 115, 19–25. [Google Scholar] [CrossRef]
- Scott, B.G.; Yang, H.; Tüzün, E.; Dong, C.; Flavell, R.A.; Christadoss, P. ICOS is essential for the development of experimental autoimmune myasthenia gravis. J. Neuroimmunol. 2004, 153, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Rojo, J.M.; Pini, E.; Ojeda, G.; Bello, R.; Dong, C.; Flavell, R.A.; Dianzani, U.; Portolés, P. CD4+ICOS+ T lymphocytes inhibit T cell activation ‘in vitro’ and attenuate autoimmune encephalitis ‘in vivo’. Int. Immunol. 2008, 20, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Elyaman, W.; Kivisäkk, P.; Reddy, J.; Chitnis, T.; Raddassi, K.; Imitola, J.; Bradshaw, E.; Kuchroo, V.K.; Yagita, H.; Sayegh, M.H.; et al. Distinct functions of autoreactive memory and effector CD4+ T cells in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2008, 173, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Hensley-McBain, T.; Heit, A.; De Rosa, S.C.; McElrath, M.J.; Andersen-Nissen, E. Optimization of a whole blood phenotyping assay for enumeration of peripheral blood leukocyte populations in multicenter clinical trials. J. Immunol. Methods 2014, 411, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Sugita, K.; Hanakawa, S.; Honda, T.; Kondoh, G.; Miyachi, Y.; Kabashima, K.; Nomura, T. Generation of Helios reporter mice and an evaluation of the suppressive capacity of Helios(+) regulatory T cells in vitro. Exp. Dermatol. 2015, 24, 554–556. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.; Martino, C.; Rahman, G.; Gonzalez, A.; Vázquez-Baeza, Y.; Mishne, G.; Knight, R. Uniform Manifold Approximation and Projection (UMAP) Reveals Composite Patterns and Resolves Visualization Artifacts in Microbiome Data. mSystems 2021, 6, e0069121. [Google Scholar] [CrossRef]
- Samusik, N.; Good, Z.; Spitzer, M.H.; Davis, K.L.; Nolan, G.P. Automated mapping of phenotype space with single-cell data. Nat. Methods 2016, 13, 493–496. [Google Scholar] [CrossRef]
- Lee, Y.H.; Thacker, R.I.; Hall, B.E.; Kong, R.; Granneman, J.G. Exploring the activated adipogenic niche: Interactions of macrophages and adipocyte progenitors. Cell Cycle 2014, 13, 184–190. [Google Scholar] [CrossRef]
- Samji, T.; Khanna, K.M. Understanding memory CD8(+) T cells. Immunol. Lett. 2017, 185, 32–39. [Google Scholar] [CrossRef]
- Marshall, H.D.; Chandele, A.; Jung, Y.W.; Meng, H.; Poholek, A.C.; Parish, I.A.; Rutishauser, R.; Cui, W.; Kleinstein, S.H.; Craft, J.; et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 2011, 35, 633–646. [Google Scholar] [CrossRef]
- Plantinga, M.; Guilliams, M.; Vanheerswynghels, M.; Deswarte, K.; Branco-Madeira, F.; Toussaint, W.; Vanhoutte, L.; Neyt, K.; Killeen, N.; Malissen, B.; et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 2013, 38, 322–335. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, J.; Yi, J.; Kim, D.; Kim, H.O.; Han, D.; Sprent, J.; Lee, Y.J.; Surh, C.D.; Cho, J.H. Phenotypic and Functional Changes of Peripheral Ly6C(+) T Regulatory Cells Driven by Conventional Effector T Cells. Front. Immunol. 2018, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Manivannan, A.; Crane, I.; Dawson, R.; Liversidge, J. Critical but divergent roles for CD62L and CD44 in directing blood monocyte trafficking in vivo during inflammation. Blood 2008, 112, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kim, C.H.; Choi, B.K.; Kwon, B.S. Origins and functional basis of regulatory CD11c+CD8+ T cells. Eur. J. Immunol. 2009, 39, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Z.; Ciric, B.; Safavi, F.; Zhang, G.X.; Rostami, A. Selective depletion of CD11c(+) CD11b(+) dendritic cells partially abrogates tolerogenic effects of intravenous MOG in murine EAE. Eur. J. Immunol. 2016, 46, 2454–2466. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Lee, G.W.; Kim, H.O.; Cho, J.H. Shaping Heterogeneity of Naive CD8(+) T Cell Pools. Immune Netw. 2023, 23, e2. [Google Scholar] [CrossRef] [PubMed]
- Coyle, A.J.; Lehar, S.; Lloyd, C.; Tian, J.; Delaney, T.; Manning, S.; Nguyen, T.; Burwell, T.; Schneider, H.; Gonzalo, J.A.; et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity 2000, 13, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- McAdam, A.J.; Chang, T.T.; Lumelsky, A.E.; Greenfield, E.A.; Boussiotis, V.A.; Duke-Cohan, J.S.; Chernova, T.; Malenkovich, N.; Jabs, C.; Kuchroo, V.K.; et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J. Immunol. 2000, 165, 5035–5040. [Google Scholar] [CrossRef]
- Tafuri, A.; Shahinian, A.; Bladt, F.; Yoshinaga, S.K.; Jordana, M.; Wakeham, A.; Boucher, L.M.; Bouchard, D.; Chan, V.S.; Duncan, G.; et al. ICOS is essential for effective T-helper-cell responses. Nature 2001, 409, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Carpenito, C.; Plesa, G.; Suhoski, M.M.; Varela-Rohena, A.; Golovina, T.N.; Carroll, R.G.; Riley, J.L.; June, C.H. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci. Transl. Med. 2010, 2, 55ra78. [Google Scholar] [CrossRef]
- Quiroga, M.F.; Pasquinelli, V.; Martínez, G.J.; Jurado, J.O.; Zorrilla, L.C.; Musella, R.M.; Abbate, E.; Sieling, P.A.; García, V.E. Inducible costimulator: A modulator of IFN-gamma production in human tuberculosis. J. Immunol. 2006, 176, 5965–5974. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Crapser, J.; Patel, A.R.; Verma, R.; Grenier, J.M.; Chauhan, A.; Jellison, E.R.; McCullough, L.D. Age-Associated Resident Memory CD8 T Cells in the Central Nervous System Are Primed To Potentiate Inflammation after Ischemic Brain Injury. J. Immunol. 2016, 196, 3318–3330. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Burmeister, Y.; Lischke, T.; Dahler, A.C.; Mages, H.W.; Lam, K.P.; Coyle, A.J.; Kroczek, R.A.; Hutloff, A. ICOS controls the pool size of effector-memory and regulatory T cells. J. Immunol. 2008, 180, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.V.; Clay, B.S.; Ferreira, C.M.; Williams, J.W.; Rogozinska, M.; Cannon, J.L.; Shilling, R.A.; Marzo, A.L.; Sperling, A.I. Protective effector memory CD4 T cells depend on ICOS for survival. PLoS ONE 2011, 6, e16529. [Google Scholar] [CrossRef]
- Simpson, T.R.; Quezada, S.A.; Allison, J.P. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr. Opin. Immunol. 2010, 22, 326–332. [Google Scholar] [CrossRef]
- Mahajan, S.; Cervera, A.; MacLeod, M.; Fillatreau, S.; Perona-Wright, G.; Meek, S.; Smith, A.; MacDonald, A.; Gray, D. The role of ICOS in the development of CD4 T cell help and the reactivation of memory T cells. Eur. J. Immunol. 2007, 37, 1796–1808. [Google Scholar] [CrossRef]
- Koutrolos, M.; Berer, K.; Kawakami, N.; Wekerle, H.; Krishnamoorthy, G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol. Commun. 2014, 2, 163. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Xiong, X.Z. ICOS(+) Tregs: A Functional Subset of Tregs in Immune Diseases. Front. Immunol. 2020, 11, 2104. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Stephens, L.A.; Anderton, S.M. Natural recovery and protection from autoimmune encephalomyelitis: Contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 2005, 175, 3025–3032. [Google Scholar] [CrossRef]
- Montero, E.; Nussbaum, G.; Kaye, J.F.; Perez, R.; Lage, A.; Ben-Nun, A.; Cohen, I.R. Regulation of experimental autoimmune encephalomyelitis by CD4+, CD25+ and CD8+ T cells: Analysis using depleting antibodies. J. Autoimmun. 2004, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Akimova, T.; Beier, U.H.; Wang, L.; Levine, M.H.; Hancock, W.W. Helios expression is a marker of T cell activation and proliferation. PLoS ONE 2011, 6, e24226. [Google Scholar] [CrossRef] [PubMed]
- Getnet, D.; Grosso, J.F.; Goldberg, M.V.; Harris, T.J.; Yen, H.R.; Bruno, T.C.; Durham, N.M.; Hipkiss, E.L.; Pyle, K.J.; Wada, S.; et al. A role for the transcription factor Helios in human CD4(+)CD25(+) regulatory T cells. Mol. Immunol. 2010, 47, 1595–1600. [Google Scholar] [CrossRef]
- Golding, A.; Hasni, S.; Illei, G.; Shevach, E.M. The percentage of FoxP3+Helios+ Treg cells correlates positively with disease activity in systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 2898–2906. [Google Scholar] [CrossRef] [PubMed]
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Döhring, C.; Samaridis, J.; Dessing, M.; Brockhaus, M.; Lanzavecchia, A.; Colonna, M. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J. Exp. Med. 1997, 185, 1743–1751. [Google Scholar] [CrossRef]
- Yi, H.; Guo, C.; Yu, X.; Zuo, D.; Wang, X.Y. Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J. Immunol. 2012, 189, 4295–4304. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, J.; Sato, K.; Nakayama, Y.; Kimura, C.; Kajino, K.; Matsui, Y.; Miyazaki, T.; Uede, T. Osteopontin modulates the generation of memory CD8+ T cells during influenza virus infection. J. Immunol. 2011, 187, 5671–5683. [Google Scholar] [CrossRef] [PubMed]
- Chabas, D.; Baranzini, S.E.; Mitchell, D.; Bernard, C.C.; Rittling, S.R.; Denhardt, D.T.; Sobel, R.A.; Lock, C.; Karpuj, M.; Pedotti, R.; et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 2001, 294, 1731–1735. [Google Scholar] [CrossRef]
- Monaghan, K.L.; Zheng, W.; Hu, G.; Wan, E.C.K. Monocytes and Monocyte-Derived Antigen-Presenting Cells Have Distinct Gene Signatures in Experimental Model of Multiple Sclerosis. Front. Immunol. 2019, 10, 2779. [Google Scholar] [CrossRef]
- MacLeod, M.; Kwakkenbos, M.J.; Crawford, A.; Brown, S.; Stockinger, B.; Schepers, K.; Schumacher, T.; Gray, D. CD4 memory T cells survive and proliferate but fail to differentiate in the absence of CD40. J. Exp. Med. 2006, 203, 897–906. [Google Scholar] [CrossRef]
- MacLeod, M.K.; Clambey, E.T.; Kappler, J.W.; Marrack, P. CD4 memory T cells: What are they and what can they do? Semin. Immunol. 2009, 21, 53–61. [Google Scholar] [CrossRef]
- MacLeod, M.K.; McKee, A.; Crawford, F.; White, J.; Kappler, J.; Marrack, P. CD4 memory T cells divide poorly in response to antigen because of their cytokine profile. Proc. Natl. Acad. Sci. USA 2008, 105, 14521–14526. [Google Scholar] [CrossRef]
- Isacke, C.M.; Horton, M.A. The Adhesion Molecule FactsBook; Academic Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Tacke, R.; Hilgendorf, I.; Garner, H.; Waterborg, C.; Park, K.; Nowyhed, H.; Hanna, R.N.; Wu, R.; Swirski, F.K.; Geissmann, F.; et al. The transcription factor NR4A1 is essential for the development of a novel macrophage subset in the thymus. Sci. Rep. 2015, 5, 10055. [Google Scholar] [CrossRef]
- Baxter, A.G. The origin and application of experimental autoimmune encephalomyelitis. Nat. Rev. Immunol. 2007, 7, 904–912. [Google Scholar] [CrossRef]
- Mangiardi, M.; Crawford, D.K.; Xia, X.; Du, S.; Simon-Freeman, R.; Voskuhl, R.R.; Tiwari-Woodruff, S.K. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol. 2011, 21, 263–278. [Google Scholar] [CrossRef]
- Mills, K.H. TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 2011, 11, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Namer, I.J.; Steibel, J.; Poulet, P.; Armspach, J.P.; Mohr, M.; Mauss, Y.; Chambron, J. Blood-brain barrier breakdown in MBP-specific T cell induced experimental allergic encephalomyelitis. A quantitative in vivo MRI study. Brain 1993, 116 Pt 1, 147–159. [Google Scholar] [CrossRef]
- Billiau, A.; Matthys, P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J. Leukoc. Biol. 2001, 70, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Lazarević, M.; Djedovic, N.; Stanisavljević, S.; Dimitrijević, M.; Stegnjaić, G.; Krishnamoorthy, G.; Mostarica Stojković, M.; Miljković, Đ.; Jevtić, B. Complete Freund’s adjuvant-free experimental autoimmune encephalomyelitis in Dark Agouti rats is a valuable tool for multiple sclerosis studies. J. Neuroimmunol. 2021, 354, 577547. [Google Scholar] [CrossRef]
- Hasselmann, J.P.C.; Karim, H.; Khalaj, A.J.; Ghosh, S.; Tiwari-Woodruff, S.K. Consistent induction of chronic experimental autoimmune encephalomyelitis in C57BL/6 mice for the longitudinal study of pathology and repair. J. Neurosci. Methods 2017, 284, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef]
- Bennett, S.A.; Roberts, D.C. Analysis of protein expression in brain tissue by ELISA. Methods Mol. Med. 2003, 79, 283–295. [Google Scholar]
- Buszko, M.; Cardini, B.; Oberhuber, R.; Oberhuber, L.; Jakic, B.; Beierfuss, A.; Wick, G.; Cappellano, G. Differential depletion of total T cells and regulatory T cells and prolonged allotransplant survival in CD3Ɛ humanized mice treated with polyclonal anti human thymocyte globulin. PLoS ONE 2017, 12, e0173088. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Markers (Phenotype) | Reference | ICOS-KO vs. C57BL/6J | ICOSL-KO vs. C57BL/6J | ICOS-KO vs. ICOSL-KO |
|---|---|---|---|---|---|
| 2 | F4/80+ CD44+ (macrophages) | [31] | x | x | ↑ |
| 3 | CD8+ CD44+ CD62L− Ly6C+ (short-lived CD8+ TEM cells) | [32] | ↑ | x | ↑ |
| 4 | CD4+ CD44+ Ly6C+ (short-lived effector CD4+T cells) | [33] | ↑ | x | ↑ |
| 5 | CD11b+ Ly6C+ CD44+ (monocytic-DC cells) | [34] | ↓ | x | ↓ |
| 8 | Ly6C+ Helios+ (peripheral Ly6C+ Tregs) | [35] | x | ↓ | x |
| 10 | CD11b+ Ly6C+ CD62L+ CD44+ (inflammatory monocytes) | [36] | x | ↑ | x |
| 14 | CD11c+ CD8+ CD44+ (CD8+ immunosuppressive Tregs) | [37] | x | x | ↑ |
| 15 | CD4+, CD11b+, CD11c+, CD44+ (tolerogenic DCs) | [38] | x | ↑ | x |
| 16 | CD3+ CD8+ Ly6C+ CD62L+ (naïve CD8+ T cells) | [39] | ↓ | x | x |
| 18 | CD3+ CD4+ CD44+ Ly6C− (long-lived CD4+ TEM cells) | [35] | ↓ | ↓ | x |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raineri, D.; Abreu, H.; Vilardo, B.; Kustrimovic, N.; Venegoni, C.; Cappellano, G.; Chiocchetti, A. Deep Flow Cytometry Unveils Distinct Immune Cell Subsets in Inducible T Cell Co-Stimulator Ligand (ICOSL)- and ICOS-Knockout Mice during Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2024, 25, 2509. https://doi.org/10.3390/ijms25052509
Raineri D, Abreu H, Vilardo B, Kustrimovic N, Venegoni C, Cappellano G, Chiocchetti A. Deep Flow Cytometry Unveils Distinct Immune Cell Subsets in Inducible T Cell Co-Stimulator Ligand (ICOSL)- and ICOS-Knockout Mice during Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2024; 25(5):2509. https://doi.org/10.3390/ijms25052509
Chicago/Turabian StyleRaineri, Davide, Hugo Abreu, Beatrice Vilardo, Natasa Kustrimovic, Chiara Venegoni, Giuseppe Cappellano, and Annalisa Chiocchetti. 2024. "Deep Flow Cytometry Unveils Distinct Immune Cell Subsets in Inducible T Cell Co-Stimulator Ligand (ICOSL)- and ICOS-Knockout Mice during Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 25, no. 5: 2509. https://doi.org/10.3390/ijms25052509
APA StyleRaineri, D., Abreu, H., Vilardo, B., Kustrimovic, N., Venegoni, C., Cappellano, G., & Chiocchetti, A. (2024). Deep Flow Cytometry Unveils Distinct Immune Cell Subsets in Inducible T Cell Co-Stimulator Ligand (ICOSL)- and ICOS-Knockout Mice during Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences, 25(5), 2509. https://doi.org/10.3390/ijms25052509