
Pterostilbene Induces Apoptosis from Endoplasmic Reticulum Stress Synergistically with Anticancer Drugs That Deposit Iron in Mitochondria


Abstract
:1. Introduction
2. Results
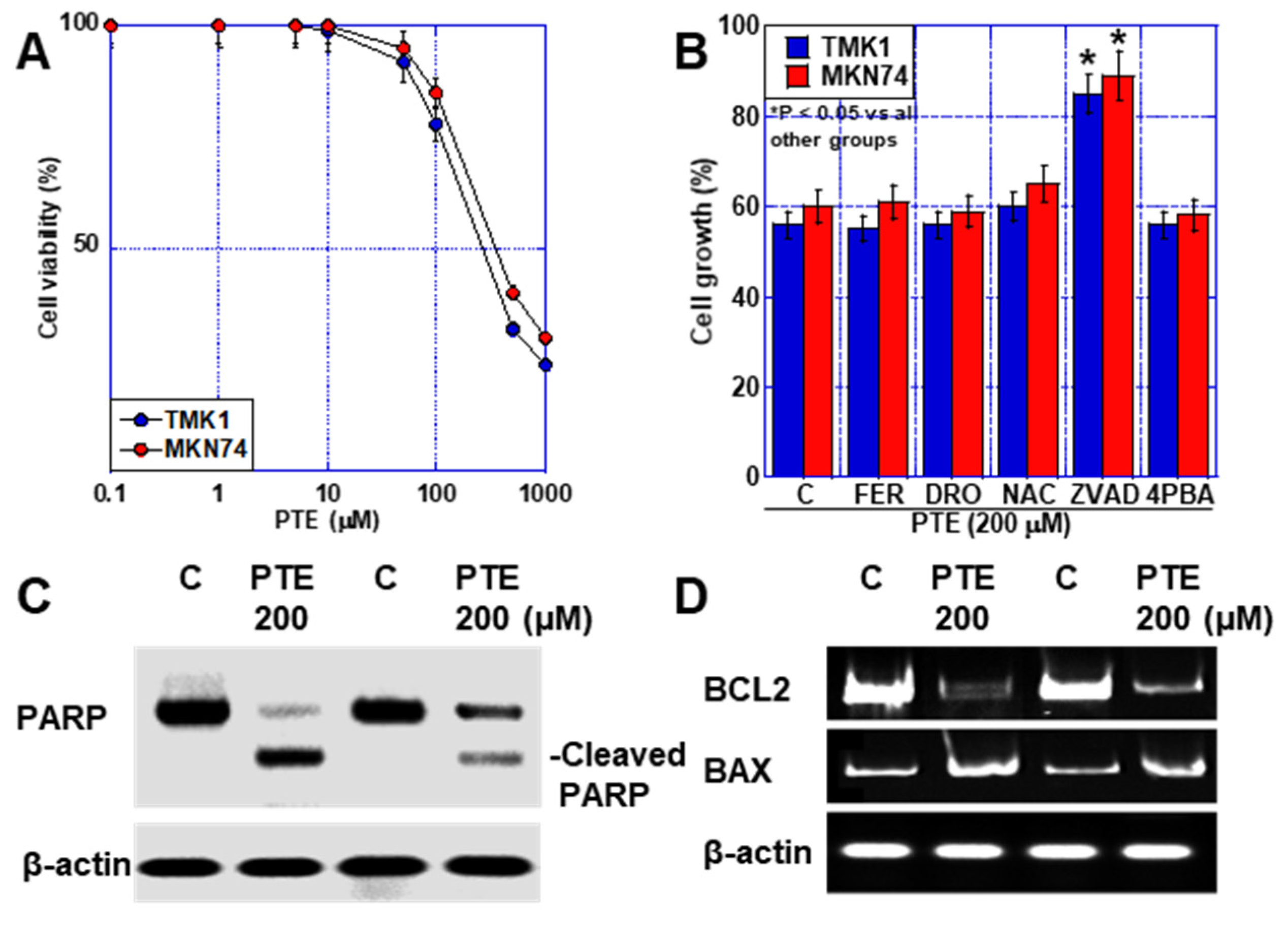
2.1. Effect of High-Dose PTE on GC Cell Lines
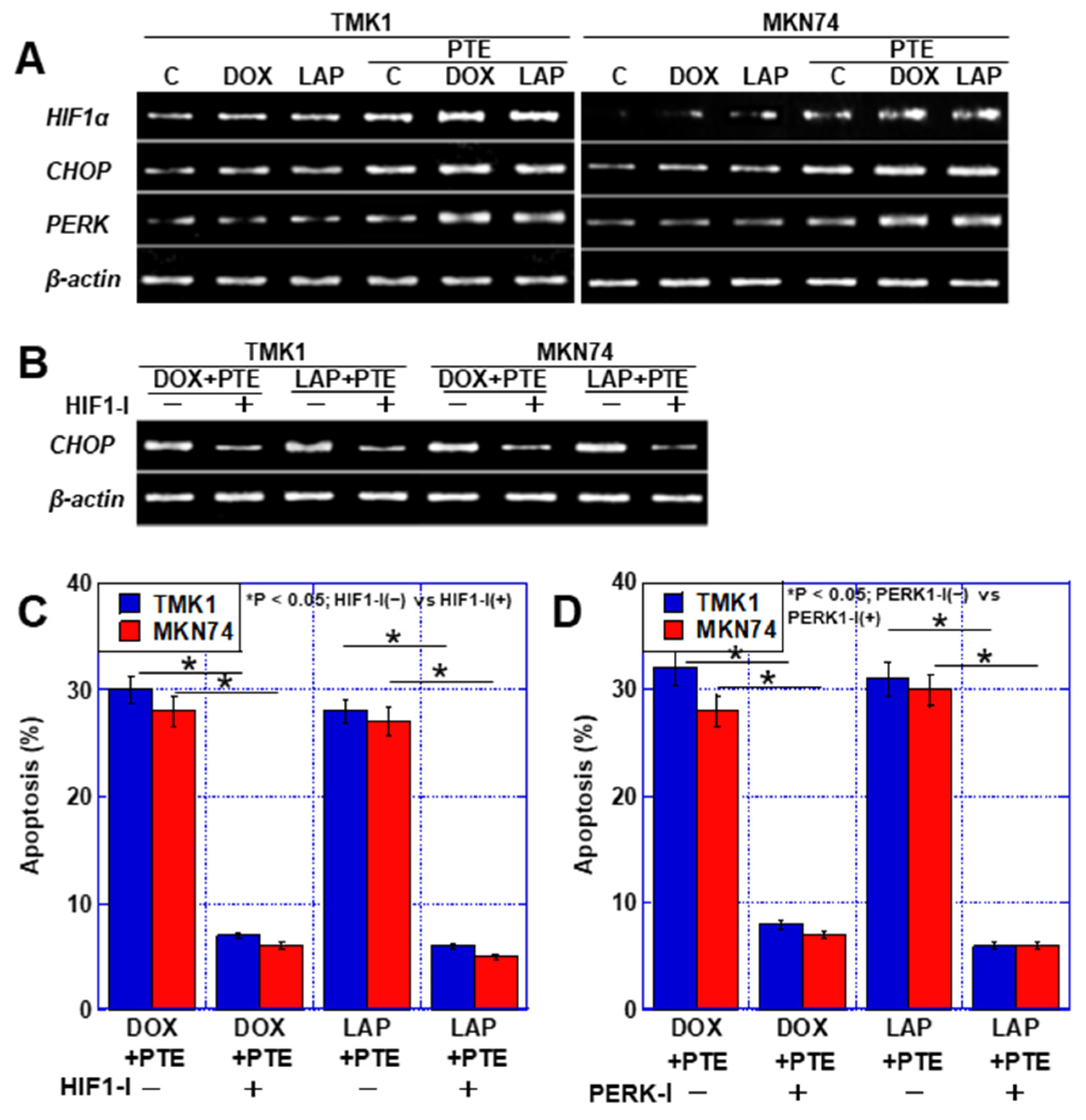
2.2. Relationship of Accumulation of mtFe with Anticancer Drugs and Sensitizing Effects of PTE
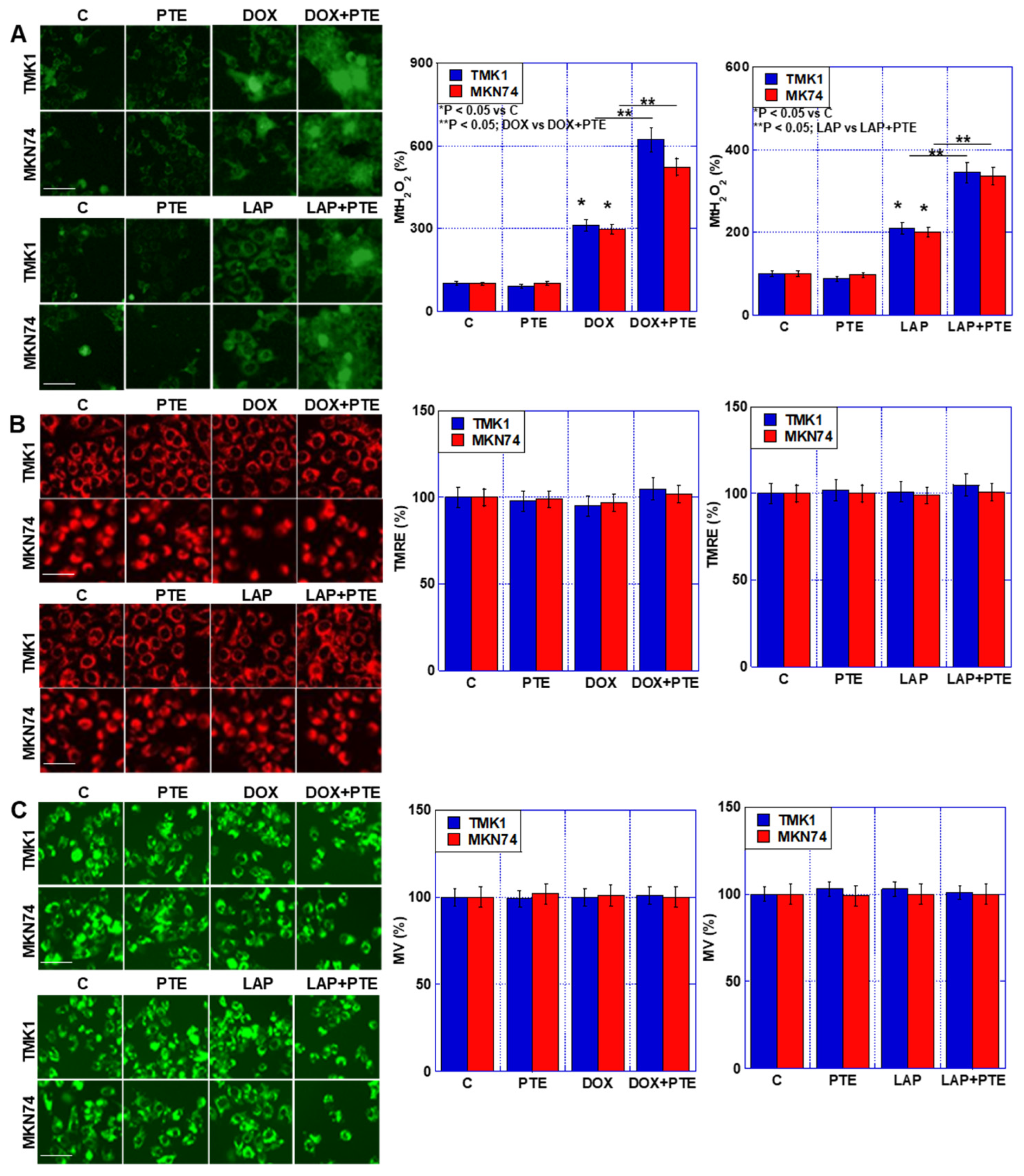
2.3. Effect of Combined Use of PTE-L and mtFe-Depositing Anticancer Drugs on Mitochondria
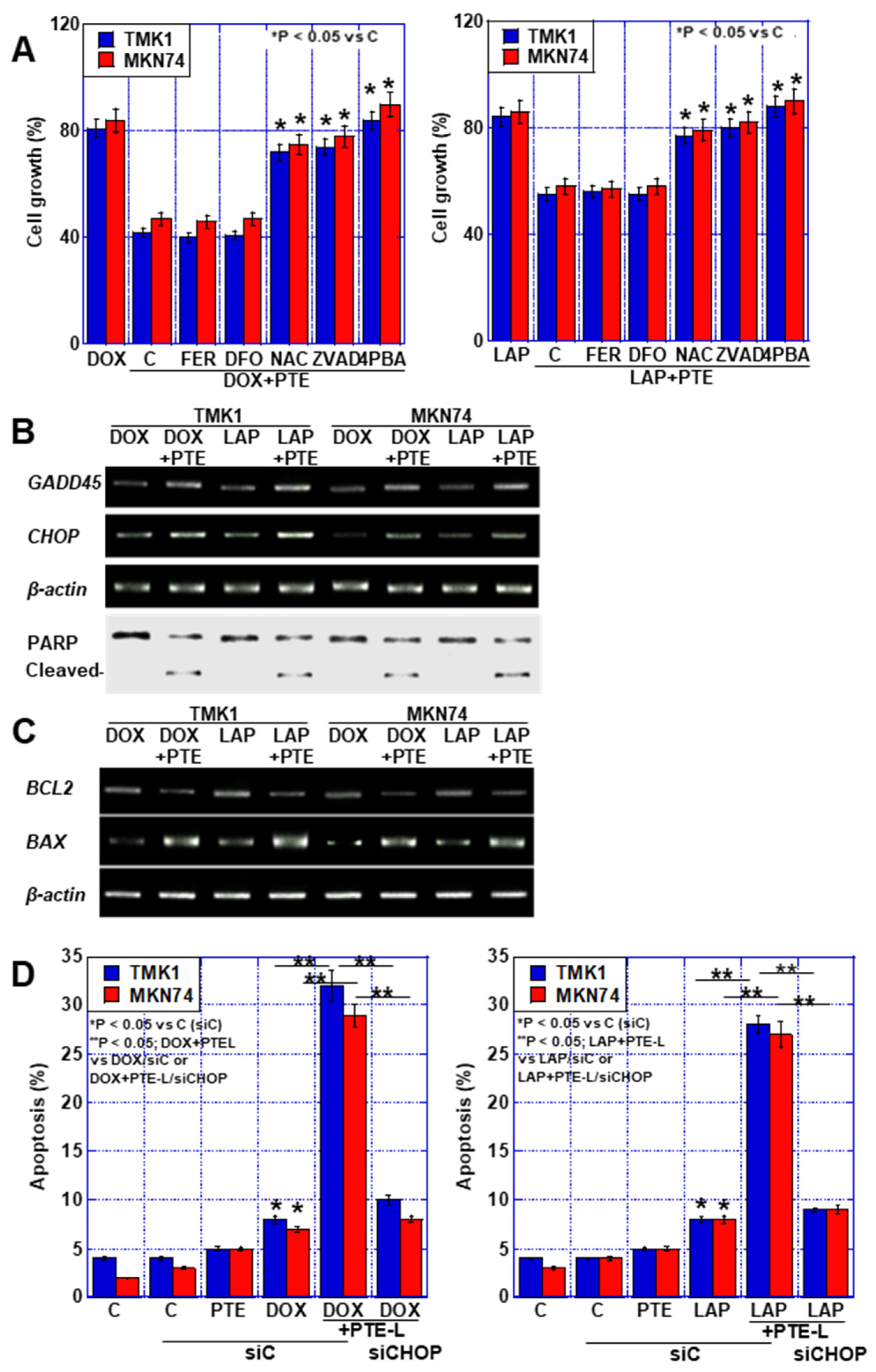
2.4. Cell Death Due to the Combination of PTE-L and mtFe-Depositing Anticancer Drugs
2.5. Synergistic Effect of PTE and Anticancer Drugs on mtFe Deposition
2.6. Effect of Combination of PTE-L and Anticancer Drugs in Mouse Subcutaneous Tumor Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Growth and Apoptosis
4.3. Reverse Transcription–Polymerase Chain Reaction
4.4. Mitochondrial Imaging
4.5. Immunoblot Analysis
4.6. Immunoprecipitation
4.7. Enzyme-Linked Immunosorbent Assay
4.8. Animals
4.9. Animal Tumor Models
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Statistics Bureau Ministry of Internal Affairs and Communications Japan. Statistical Handbook of Japan 2017; Statistics Bureau Ministry of Internal Affairs and Communications Japan: Tokyo, Japan, 2017. Available online: https://www.stat.go.jp/english/data/handbook/pdf/2017all.pdf (accessed on 7 December 2023).
- International Agency for Research on Cancer. Globocan 2018 Latest Global Cancer Data. Available online: https://www.iarc.who.int/infographics/globocan-2018-latest-global-cancer-data/ (accessed on 7 December 2023).
- Mikami, H.; Nagase, H. Cancer Survival Rates at Japanese Association of Clinical Cancer Centers. Available online: https://kapweb.chiba-cancer-registry.org/ (accessed on 8 December 2023).
- Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011, 14, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e13. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Yang, Y.S.; Xu, D.P.; Qu, J.H.; Guo, M.Z.; Gong, Y.; Huang, J. Comparative study on overexpression of HER2/neu and HER3 in gastric cancer. World J. Surg. 2009, 33, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.; Tollefsbol, T.O. Pterostilbene down-regulates hTERT at physiological concentrations in breast cancer cells: Potentially through the inhibition of cMyc. J. Cell Biochem. 2018, 119, 3326–3337. [Google Scholar] [CrossRef] [PubMed]
- Hong Bin, W.; Da, L.H.; Xue, Y.; Jing, B. Pterostilbene (3′,5′-dimethoxy-resveratrol) exerts potent antitumor effects in HeLa human cervical cancer cells via disruption of mitochondrial membrane potential, apoptosis induction and targeting m-TOR/PI3K/Akt signalling pathway. J. BUON 2018, 23, 1384–1389. [Google Scholar] [PubMed]
- Moon, D.; McCormack, D.; McDonald, D.; McFadden, D. Pterostilbene induces mitochondrially derived apoptosis in breast cancer cells in vitro. J. Surg. Res. 2013, 180, 208–215. [Google Scholar] [CrossRef]
- Mori, S.; Kishi, S.; Honoki, K.; Fujiwara-Tani, R.; Moriguchi, T.; Sasaki, T.; Fujii, K.; Tsukamoto, S.; Fujii, H.; Kido, A.; et al. Anti-Stem Cell Property of Pterostilbene in Gastrointestinal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9347. [Google Scholar] [CrossRef]
- Hojo, Y.; Kishi, S.; Mori, S.; Fujiwara-Tani, R.; Sasaki, T.; Fujii, K.; Nishiguchi, Y.; Nakashima, C.; Luo, Y.; Shinohara, H.; et al. Sunitinib and Pterostilbene Combination Treatment Exerts Antitumor Effects in Gastric Cancer via Suppression of PDZD8. Int. J. Mol. Sci. 2022, 23, 4002. [Google Scholar] [CrossRef]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knüchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef]
- Wang, R.; Hussain, A.; Guo, Q.Q.; Jin, X.W.; Wang, M.M. Oxygen and Iron Availability Shapes Metabolic Adaptations of Cancer Cells. World J. Oncol. 2024, 15, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.T.; Chen, P.W.; Li, S.; Ke, T.M.; Lin, S.H.; Yang, C.C. Pterostilbene inhibits lung squamous cell carcinoma growth in vitro and in vivo by inducing S phase arrest and apoptosis. Oncol. Lett. 2019, 18, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Elsherbini, A.M.; Sheweita, S.A.; Sultan, A.S. Pterostilbene as a Phytochemical Compound Induces Signaling Pathways Involved in the Apoptosis and Death of Mutant P53-Breast Cancer Cell Lines. Nutr. Cancer 2021, 73, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Han, J.M.; Choi, Y.S.; Jung, H.J. Pterostilbene Suppresses both Cancer Cells and Cancer Stem-Like Cells in Cervical Cancer with Superior Bioavailability to Resveratrol. Molecules 2020, 25, 228. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, Z.; Xu, W.; Wang, Q.; Zhang, C.; Ding, Y.; Nie, W.; Lai, J.; Chen, Y.; Huang, H. Pterostilbene promotes mitochondrial apoptosis and inhibits proliferation in glioma cells. Sci. Rep. 2021, 11, 6381. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Chen, G.; Xu, Z.; Yang, G.; Li, B.; Wu, X.; Xiao, W.; Xie, B.; Hu, L.; Sun, X.; et al. Pterostilbene induces apoptosis and cell cycle arrest in diffuse large B-cell lymphoma cells. Sci. Rep. 2016, 6, 37417. [Google Scholar] [CrossRef]
- Chen, R.J.; Lyu, Y.J.; Chen, Y.Y.; Lee, Y.C.; Pan, M.H.; Ho, Y.S.; Wang, Y.J. Chloroquine Potentiates the Anticancer Effect of Pterostilbene on Pancreatic Cancer by Inhibiting Autophagy and Downregulating the RAGE/STAT3 Pathway. Molecules 2021, 26, 6741. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, J.; Chu, Q.Z.; Ji, W.K. PDZD8-mediated lipid transfer at contacts between the ER and late endosomes/lysosomes is required for neurite outgrowth. J. Cell Sci. 2022, 135, jcs255026. [Google Scholar] [CrossRef]
- Lipper, C.H.; Stofleth, J.T.; Bai, F.; Sohn, Y.S.; Roy, S.; Mittler, R.; Nechushtai, R.; Onuchic, J.N.; Jennings, P.A. Redox-dependent gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. USA 2019, 116, 19924–19929. [Google Scholar] [CrossRef]
- Tan, G.; Liu, D.; Pan, F.; Zhao, J.; Li, T.; Ma, Y.; Shen, B.; Lyu, J. His-87 ligand in mitoNEET is crucial for the transfer of iron sulfur clusters from mitochondria to cytosolic aconitase. Biochem. Biophys. Res. Commun. 2016, 470, 226–232. [Google Scholar] [CrossRef]
- Chang, H.C.; Shapiro, J.S.; Jiang, X.; Senyei, G.; Sato, T.; Geier, J.; Sawicki, K.T.; Ardehali, H. Augmenter of liver regeneration regulates cellular iron homeostasis by modulating mitochondrial transport of ATP-binding cassette B8. eLife 2021, 10, e65158. [Google Scholar] [CrossRef] [PubMed]
- Issitt, T.; Bosseboeuf, E.; De Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell 2010, 21, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Emerling, B.M.; Platanias, L.C.; Black, E.; Nebreda, A.R.; Davis, R.J.; Chandel, N.S. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell Biol. 2005, 25, 4853–4862. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, R.; Xia, T.; Hsu, E.; Cai, Y.; Gu, Z.; Hankinson, O. Inhibitory effects of nitric oxide on invasion of human cancer cells. Cancer Lett. 2007, 257, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Berchner-Pfannschmidt, U.; Möller, U.; Brecht, M.; Wotzlaw, C.; Acker, H.; Jungermann, K.; Kietzmann, T. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 4302–4307. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Verras, M.; McNeil, B.; Koong, A.C.; Denko, N.C. Plant stilbenes induce endoplasmic reticulum stress and their anti-cancer activity can be enhanced by inhibitors of autophagy. Exp. Cell Res. 2015, 339, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, X.Q.; Chen, H.Y.; Liu, B.H. Involvement of the Nrf2 pathway in the regulation of pterostilbene-induced apoptosis in HeLa cells via ER stress. J. Pharmacol. Sci. 2014, 126, 216–229. [Google Scholar] [CrossRef]
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. HIF-1α triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci. Rep. 2018, 8, 17939. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Michel, S.; Canonne, M.; Arnould, T.; Renard, P. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 2015, 21, 58–68. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T. Adriamycin (doxorubicin) cardiotoxicity: A review. West. J. Med. 1979, 131, 364–368. [Google Scholar] [PubMed]
- Ochiai, A.; Yasui, W.; Tahara, E. Growth-promoting effect of gastrin on human gastric carcinoma cell line TMK-1. Jpn. J. Cancer Res. 1985, 76, 1064–1071. [Google Scholar] [PubMed]
- Motoyama, T.; Hojo, H.; Watanabe, H. Comparison of seven cell lines derived from human gastric carcinomas. Acta Pathol. Jpn. 1986, 36, 65–83. [Google Scholar] [CrossRef]
- Yokozaki, H. Molecular characteristics of eight gastric cancer cell lines established in Japan. Pathol. Int. 2000, 50, 767–777. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Yasui, W.; Pettaway, C.A.; Yano, S.; Oue, N.; Tahara, E.; Fidler, I.J. Interferon-alpha prevents selection of doxorubicin-resistant undifferentiated-androgen-insensitive metastatic human prostate cancer cells. Prostate 2001, 49, 19–29. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Set | |||
|---|---|---|---|
| Gene symbol | Gene bank ID | Forward primer (5′–3′) | Reverse primer (5′–3′) |
| BCL2 | M13994.1 | acgacaaccgggagatagtg | catcccagcctccgttatcc |
| BAX | L22473.1 | catgaagacaggggcccttt | cttccagatggtgagcgagg |
| GADD45 | M60974.1 | ggaggaattctcggctggag | tccatgtagcgactttcccg |
| CHOP | NM_001195053.1 | ccagccactccccattatcc | ttcggtcaatcagagctcgg |
| PDZD8 | NM_173791.4 | tcctcgtgttgatgctgaag | ttgtctgacgtgttgggtgt |
| mitoNEET (CISD1) | BC007043.1 | tccagaaagacaaccccaag | gcccacattgtctccagtct |
| ABCB8 | NM_001282291.2 | cgtggggtctcgctttaact | cctgacactggcgagacaat |
| HIF1α | AF208487.1 | gaaagcgcaagtcctcaaag | tgggtaggagatggagatgc |
| PERK | NM_004836.7 | gcagaggcagtggagtttct | ggcaaagggctatgggagtt |
| ACTB | NM_001101.3 | ggacttcgagcaagagatgg | agcactgtgttggcgtacag |
| Antibody | |||
| Protein | Clone | Company | |
| PARP | - | GeneTex, Irvine, CA, USA | |
| mitoNEET | L70G2 | Biocompare, South San Francisco, CA, USA | |
| ABCB8 | F-4 | Santa Cruz, Santa Cruz, CA, USA | |
| PDZD8 | - | Bioss Inc, Woburn, MA, USA | |
| β-actin | - | Abcam, Cambridge, MA, USA | |
| ELISA | |||
| Items | Catalog number | Company | |
| Hydrogen peroxide | 21024 | Aoxre Bioscience, Burlingane, CA, USA | |
| Human CHOP | LS-F8872 | Lsbio, Shirley, MA, USA | |
| Human HIF1α | EHIF1A | Thermo Fisher, Tokyo, Japan | |
| 4HNE | ab238538 | Abcam, Cambridge, MA, USA | |
| Human GPX4 | ARP-E4145 | Biocompare. South San Francisco, CA, USA | |
| Cleaved CK18, M30 | 10011 | VLVbio, Nacka, Sweden | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishiguch, Y.; Fujiwara-Tani, R.; Nukaga, S.; Nishida, R.; Ikemoto, A.; Sasaki, R.; Mori, S.; Ogata, R.; Kishi, S.; Hojo, Y.; et al. Pterostilbene Induces Apoptosis from Endoplasmic Reticulum Stress Synergistically with Anticancer Drugs That Deposit Iron in Mitochondria. Int. J. Mol. Sci. 2024, 25, 2611. https://doi.org/10.3390/ijms25052611
Nishiguch Y, Fujiwara-Tani R, Nukaga S, Nishida R, Ikemoto A, Sasaki R, Mori S, Ogata R, Kishi S, Hojo Y, et al. Pterostilbene Induces Apoptosis from Endoplasmic Reticulum Stress Synergistically with Anticancer Drugs That Deposit Iron in Mitochondria. International Journal of Molecular Sciences. 2024; 25(5):2611. https://doi.org/10.3390/ijms25052611
Chicago/Turabian StyleNishiguch, Yukiko, Rina Fujiwara-Tani, Shota Nukaga, Ryoichi Nishida, Ayaka Ikemoto, Rika Sasaki, Shiori Mori, Ruiko Ogata, Shingo Kishi, Yudai Hojo, and et al. 2024. "Pterostilbene Induces Apoptosis from Endoplasmic Reticulum Stress Synergistically with Anticancer Drugs That Deposit Iron in Mitochondria" International Journal of Molecular Sciences 25, no. 5: 2611. https://doi.org/10.3390/ijms25052611
APA StyleNishiguch, Y., Fujiwara-Tani, R., Nukaga, S., Nishida, R., Ikemoto, A., Sasaki, R., Mori, S., Ogata, R., Kishi, S., Hojo, Y., Shinohara, H., Sho, M., & Kuniyasu, H. (2024). Pterostilbene Induces Apoptosis from Endoplasmic Reticulum Stress Synergistically with Anticancer Drugs That Deposit Iron in Mitochondria. International Journal of Molecular Sciences, 25(5), 2611. https://doi.org/10.3390/ijms25052611