
FlashPCR: Revolutionising qPCR by Accelerating Amplification through Low ∆T Protocols


Abstract
:1. Introduction
2. Results
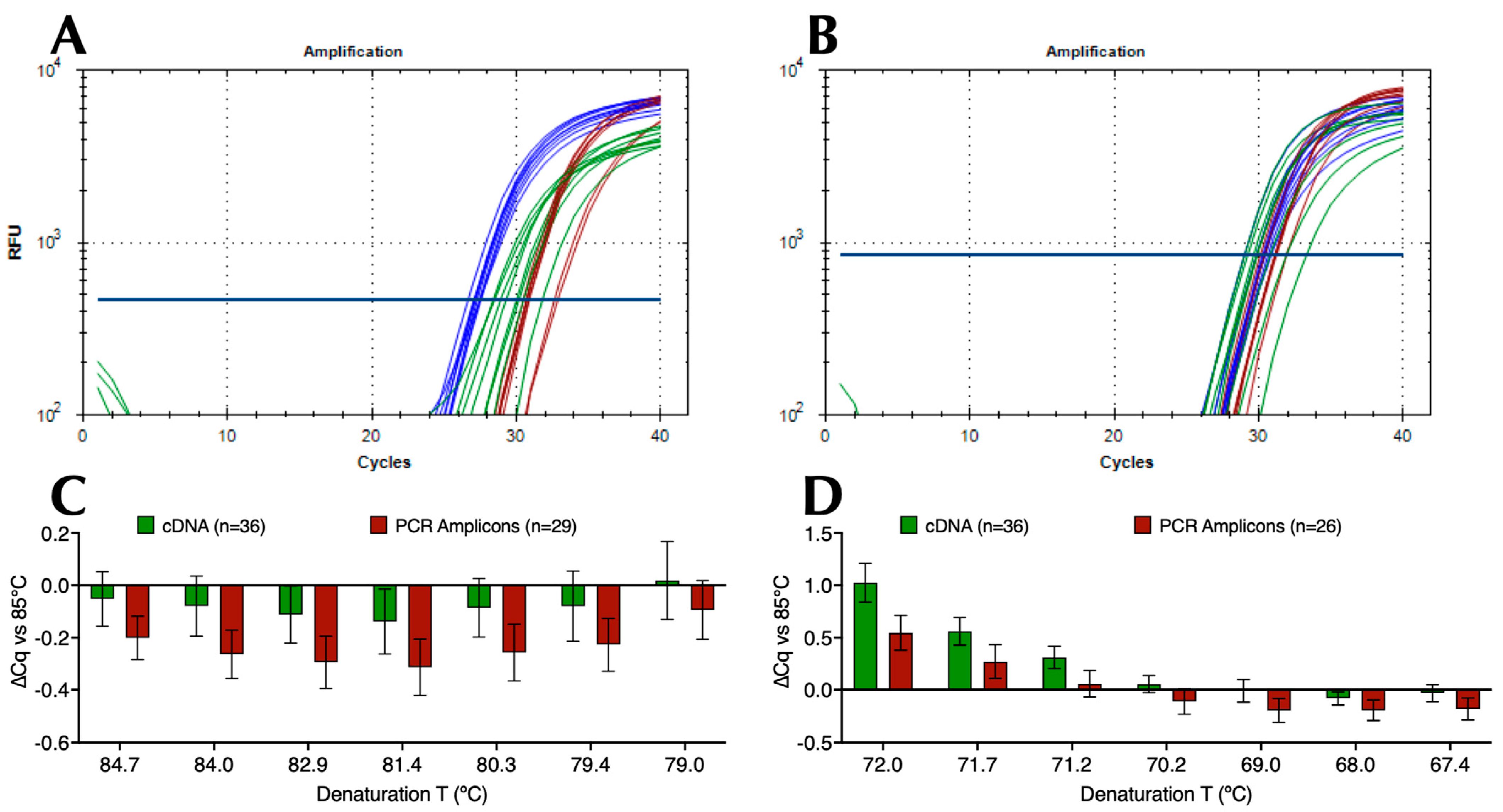
2.1. Amplification across Denaturation and Polymerisation Gradients
2.2. Amplification Using Single Denaturation and Polymerisation Temperatures
2.3. Amplification with Short Primers Modified with Pentabases
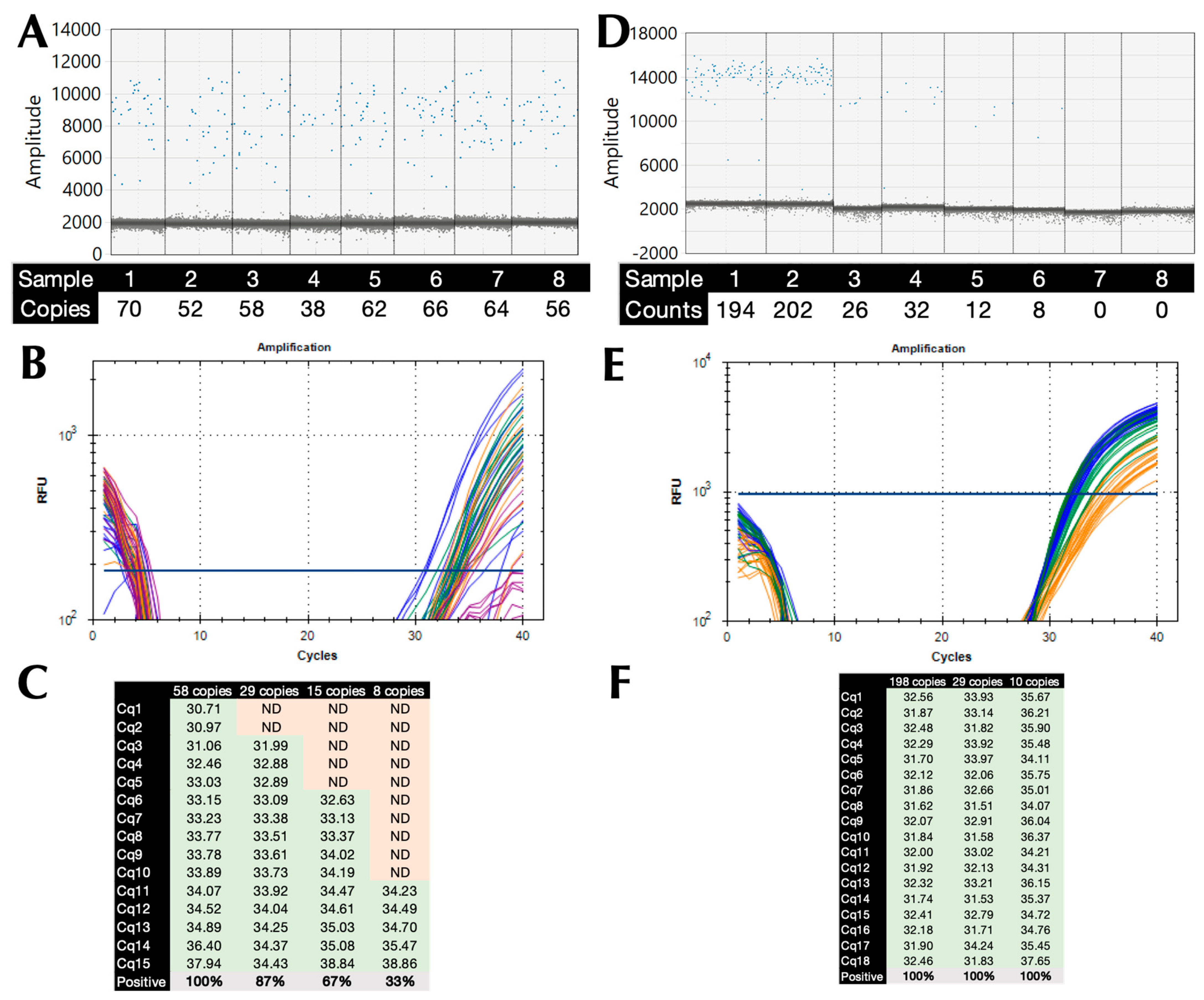
2.4. Limit of Detection (LoD) and PCR Efficiency
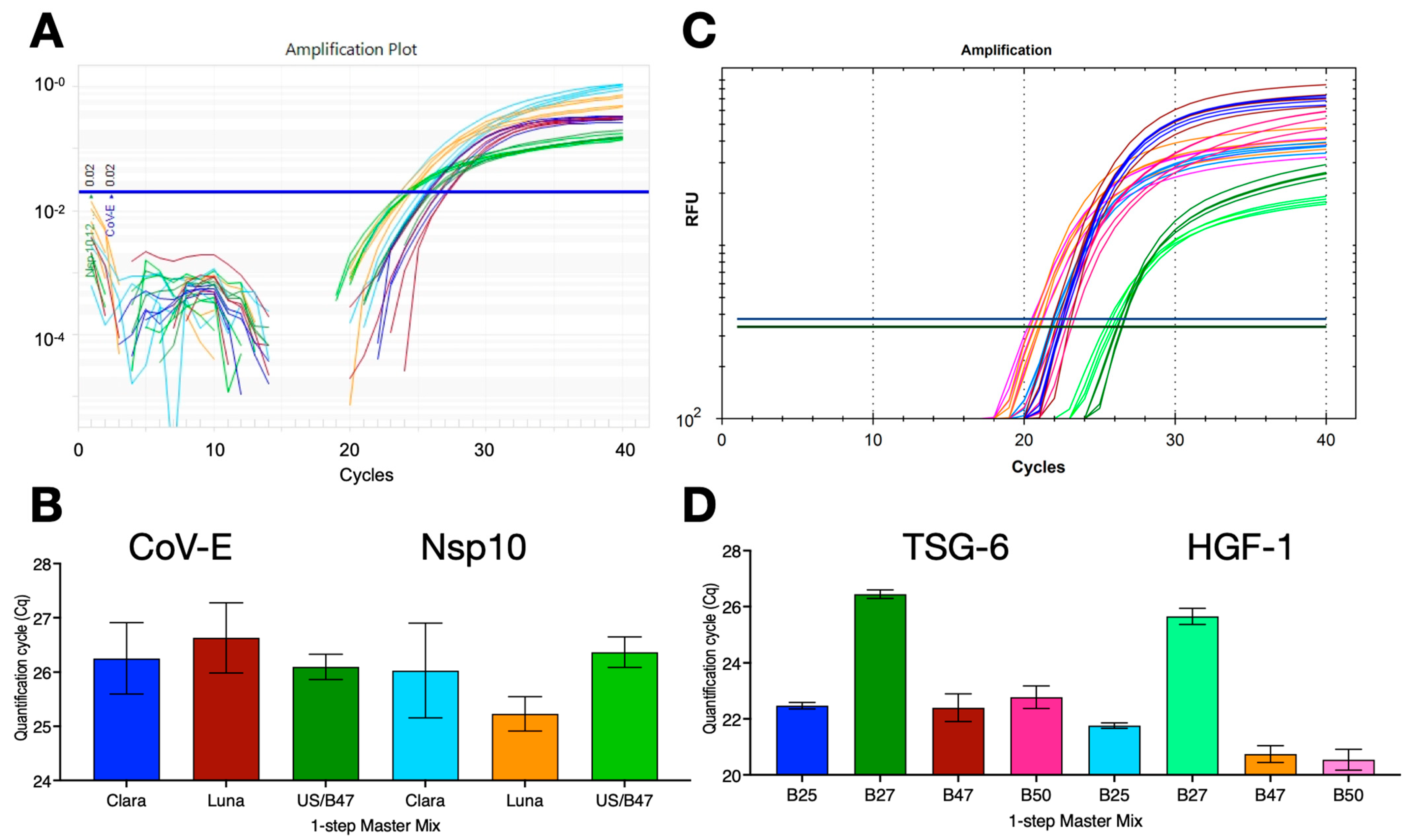
2.5. Other Pathogens
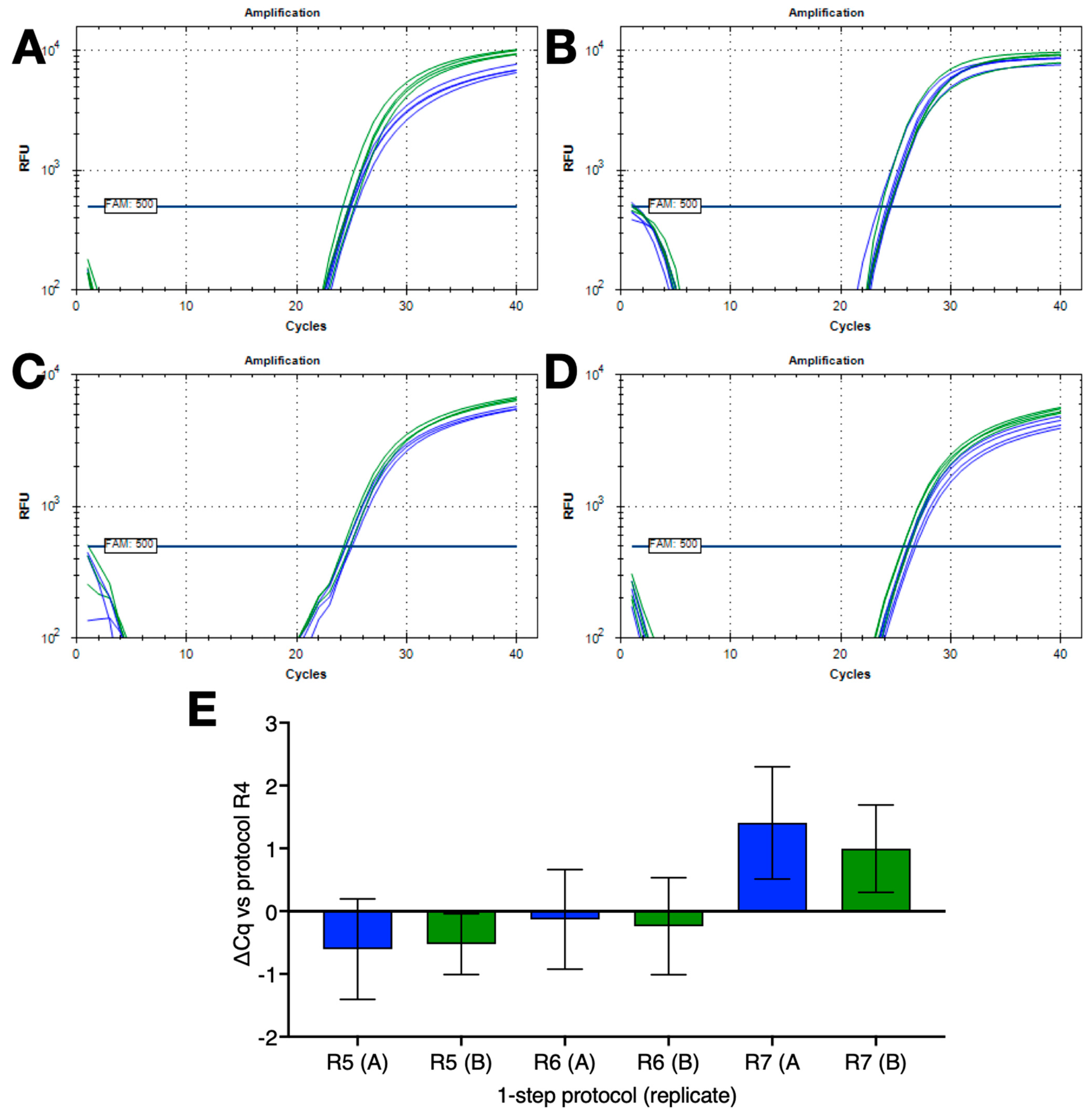
2.6. RT and PCR
3. Discussion
- Primer designs that incorporate Tms higher than conventionally recommended;
- Modifications such as Pentabases that allow the use of short primers whilst maintaining a high Tm;
- Flexible probe designs that tolerate overlap of the 3′-ends of probes with 3′-ends of primers binding to opposite strands;
- Simple buffers that balance the requirement to denature PCR amplicons with the ability of primers to hybridise and prime polymerisation;
- Low ∆T protocols that involve running qPCR reactions at denaturation temperatures of approximately 80 °C and polymerization temperatures around 70 °C;
- Short cycling times that minimise qPCR run times;
- Wide applicability demonstrated through the targeting of genomic DNA from various common pathogens;
- Efficient one- and two-step RT-qPCR amplification of viral gRNA and cellular mRNA, even in the absence of a dedicated RT step.
4. Materials and Methods
4.1. Reagents and qPCR Instruments
4.2. Primers and Probes
4.3. RNA Extractions
4.4. cDNA Synthesis
4.4.1. SARS-CoV-2
4.4.2. Human RNA
4.5. qPCR Reactions
4.6. ddPCR Reactions
4.7. Pathogen DNA
4.8. 1-Step RT-qPCR Reactions
4.9. Data Analysis
5. Conclusions
6. Note Added in Proof
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Higuchi, R.; Dollinger, G.; Walsh, P.S.; Griffith, R. Simultaneous amplification and detection of specific DNA sequences. Bio/Technology 1992, 10, 413–417. [Google Scholar] [CrossRef]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Bio/Technology 1993, 11, 1026–1030. [Google Scholar]
- Bustin, S.A.; Mueller, R. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin. Sci. 2005, 109, 365–379. [Google Scholar] [CrossRef]
- Bustin, S.; Penning, L.C. Improving the analysis of quantitative PCR data in veterinary research. Vet. J. 2012, 191, 279–281. [Google Scholar] [CrossRef]
- Lopez, M.M.; Llop, P.; Olmos, A.; Marco-Noales, E.; Cambra, M.; Bertolini, E. Are molecular tools solving the challenges posed by detection of plant pathogenic bacteria and viruses? Curr. Issues Mol. Biol. 2009, 11, 13–46. [Google Scholar]
- Abramova, A.; Berendonk, T.U.; Bengtsson-Palme, J. A global baseline for qPCR-determined antimicrobial resistance gene prevalence across envi-ronments. Environ. Int. 2023, 178, 108084. [Google Scholar] [CrossRef]
- Khot, P.D.; Fredricks, D.N. PCR-based diagnosis of human fungal infections. Expert Rev. Anti-Infect. Ther. 2009, 7, 1201–1221. [Google Scholar] [CrossRef] [PubMed]
- Nouwairi, R.L.; Cunha, L.L.; Turiello, R.; Scott, O.; Hickey, J.; Thomson, S.; Knowles, S.; Chapman, J.D.; Landers, J.P. Ultra-rapid real-time microfluidic RT-PCR instrument for nucleic acid analysis. Lab Chip 2022, 22, 3424–3435. [Google Scholar] [CrossRef]
- Dahl, A.; Sultan, M.; Jung, A.; Schwartz, R.; Lange, M.; Steinwand, M.; Livak, K.J.; Lehrach, H.; Nyarsik, L. Quantitative PCR based expression analysis on a nanoliter scale using polymer nano-well chips. Biomed. Microdevices 2007, 9, 307–314. [Google Scholar] [CrossRef]
- Qiu, X.; Ge, S.; Gao, P.; Li, K.; Yang, Y.; Zhang, S.; Ye, X.; Xia, N.; Qian, S. A Low-Cost and Fast Real-Time PCR System Based on Capillary Convection. SLAS Technol. 2017, 22, 13–17. [Google Scholar] [CrossRef]
- Fernández-Carballo, B.L.; McGuiness, I.; McBeth, C.; Kalashnikov, M.; Borrós, S.; Sharon, A.; Sauer-Budge, A.F. Low-cost, real-time, continuous flow PCR system for pathogen detection. Biomed. Microdevices 2016, 18, 34. [Google Scholar] [CrossRef]
- Mohammadyousef, P.; Paliouras, M.; Trifiro, M.A.; Kirk, A.G. Plasmonic and label-free real-time quantitative PCR for point-of-care diagnostics. Analyst 2021, 146, 5619–5630. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, N.R.; Bolene, M.A.E.; Jaspan, M.; Ayers, A.G.; Zarrandikoetxea, S.; Freudman, J.; Shah, N.; Tolwani, A.M.; Hu, Y.; Chern, T.L.; et al. Multiplexed reverse-transcriptase quantitative polymerase chain reaction using plasmonic nanoparticles for point-of-care COVID-19 diagnosis. Nat. Nanotechnol. 2022, 17, 984–992. [Google Scholar] [CrossRef]
- Wittwer, C.T. Rapid Cycle and Extreme Polymerase Chain Reaction. Methods Mol. Biol. 2023, 2621, 257–266. [Google Scholar]
- Millington, A.L.; Houskeeper, J.A.; Quackenbush, J.F.; Trauba, J.M.; Wittwer, C.T. The kinetic requirements of extreme qPCR. Biomol. Detect. Quantif. 2019, 17, 100081. [Google Scholar] [CrossRef]
- Farrar, J.S.; Wittwer, C.T. Extreme PCR: Efficient and Specific DNA Amplification in 15–60 Seconds. Clin. Chem. 2015, 61, 145–153. [Google Scholar] [CrossRef]
- Paruch, L. Molecular Diagnostic Tools Applied for Assessing Microbial Water Quality. Int. J. Environ. Res. Public Health 2022, 19, 5128. [Google Scholar] [CrossRef] [PubMed]
- Foddai, A.C.G.; Grant, I.R. Methods for detection of viable foodborne pathogens: Current state-of-art and future prospects. Appl. Microbiol. Biotechnol. 2020, 104, 4281–4288. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; McCord, B.; Buel, E. Advances in forensic DNA quantification: A review. Electrophoresis 2014, 35, 3044–3052. [Google Scholar] [CrossRef]
- Henry, R.; Schang, C.; Chandrasena, G.I.; Deletic, A.; Edmunds, M.; Jovanovic, D.; Kolotelo, P.; Schmidt, J.; Williamson, R.; McCarthy, D. Environmental monitoring of waterborne Campylobacter: Evaluation of the Australian standard and a hybrid extraction-free MPN-PCR method. Front. Microbiol. 2015, 6, 74. [Google Scholar] [CrossRef]
- Tagliavia, M.; Nicosia, A.; Salamone, M.; Biondo, G.; Bennici, C.D.; Mazzola, S.; Cuttitta, A. Development of a fast DNA extraction method for sea food and marine species identification. Food Chem. 2016, 203, 375–378. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.; Lv, M.; Sun, Y.; Cao, J. Clinical Validation of DNA Extraction-Free qPCR, Visual LAMP, and Fluorescent LAMP Assays for the Rapid Detection of African Swine Fever Virus. Life 2022, 12, 1067. [Google Scholar] [CrossRef]
- Ratti, C.; Minguzzi, S.; Turina, M. A Rapid Protocol of Crude RNA/DNA Extraction for RT-qPCR Detection and Quantification. Methods Mol. Biol. 2019, 1875, 159–169. [Google Scholar]
- Minguzzi, S.; Terlizzi, F.; Lanzoni, C.; Poggi Pollini, C.; Ratti, C. A Rapid Protocol of Crude RNA/DNA Extraction for RT-qPCR Detection and Quantification of ‘Candidatus Phytoplasma prunorum’. PLoS ONE 2016, 11, e0146515. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Ghosh, A. Rapid and zero-cost DNA extraction from soft-bodied insects for routine PCR-based applications. PLoS ONE 2022, 17, e0271312. [Google Scholar] [CrossRef]
- Delgado-Diaz, D.J.; Sakthivel, D.; Nguyen, H.H.T.; Farrokzhad, K.; Hopper, W.; Narh, C.A.; Richards, J.S. Strategies That Facilitate Extraction-Free SARS-CoV-2 Nucleic Acid Amplification Tests. Viruses 2022, 14, 1311. [Google Scholar] [CrossRef]
- Bustin, S.; Coward, A.; Sadler, G.; Teare, L.; Nolan, T. CoV2-ID, a MIQE-compliant sub-20-min 5-plex RT-PCR assay targeting SARS-CoV-2 for the diagnosis of COVID-19. Sci. Rep. 2020, 10, 22214. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Shipley, G.L.; Kirvell, S.; Mueller, R.; Nolan, T. RT-qPCR Detection of SARS-CoV-2: No Need for a Dedicated Reverse Transcription Step. Int. J. Mol. Sci. 2022, 23, 1303. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Forootan, A.; Sjöback, R.; Björkman, J.; Sjögreen, B.; Linz, L.; Kubista, M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 2017, 12, 1–6. [Google Scholar] [CrossRef]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Fan, H.; Wang, J.; Komiyama, M.; Liang, X. Effects of secondary structures of DNA templates on the quantification of qPCR. J Biomol Struct Dyn. 2019, 37, 2867–2874. [Google Scholar] [CrossRef] [PubMed]
- Gavina, K.; Franco, L.C.; Khan, H.; Lavik, J.P.; Relich, R.F. Molecular point-of-care devices for the diagnosis of infectious diseases in resource-limited set-tings—A review of the current landscape, technical challenges, and clinical impact. J. Clin. Virol. 2023, 169, 105613. [Google Scholar] [CrossRef]
- Yadav, S.K.; Verma, D.; Yadav, U.; Kalkal, A.; Priyadarshini, N.; Kumar, A.; Mahato, K. Point-of-Care Devices for Viral Detection: COVID-19 Pandemic and Beyond. Micromachines 2023, 14, 1744. [Google Scholar] [CrossRef] [PubMed]
- Heidt, B.; Siqueira, W.F.; Eersels, K.; Diliën, H.; van Grinsven, B.; Fujiwara, R.T.; Cleij, T.J. Point of Care Diagnostics in Resource-Limited Settings: A Review of the Present and Future of PoC in Its Most Needed Environment. Biosensors 2020, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Hilscher, C.; Vahrson, W.; Dittmer, D.P. Faster quantitative real-time PCR protocols may lose sensitivity and show increased variability. Nucleic Acids Res. 2005, 33, e182. [Google Scholar] [CrossRef] [PubMed]
- García-Bernalt Diego, J.; Fernández-Soto, P.; Muro, A. The Future of Point-of-Care Nucleic Acid Amplification Diagnostics after COVID-19: Time to Walk the Walk. Int. J. Mol. Sci. 2022, 23, 14110. [Google Scholar] [CrossRef] [PubMed]
- Calorenni, P.; Leonardi, A.A.; Sciuto, E.L.; Rizzo, M.G.; Faro, M.J.L.; Fazio, B.; Irrera, A.; Conoci, S. PCR-Free Innovative Strategies for SARS-CoV-2 Detection. Adv. Healthc. Mater. 2023, 12, e2300512. [Google Scholar] [CrossRef]
- Frey, U.H.; Bachmann, H.S.; Peters, J.; Siffert, W. PCR-amplification of GC-rich regions: ‘slowdown PCR’. Nat Protoc. 2008, 3, 1312–1317. [Google Scholar] [CrossRef]
- Jin, J.; Lee, Y.K.; Wickes, B.L. Simple chemical extraction method for DNA isolation from Aspergillus fumigatus and other Aspergillus species. J. Clin. Microbiol. 2004, 42, 4293–4296. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Supplier | Reagent/Instrument/Software | Part No. |
|---|---|---|
| ABI, Warrington, UK | TaqMan Fast Advanced Master Mix | 4444553 |
| Agilent, Stockport, UK | 2100 Bioanalyzer | G2939BA |
| Bioline, London, UK | SensiFast SYBR No-ROX | BIO-98050 |
| Bioline, London, UK | MyTaq DNA polymerase | BIO-21105 |
| Bioline, London, UK | SensiFast Probe No-ROX | BIO-86050 |
| Biomolecular Systems, London, UK | Mic qPCR Cycler | N/A |
| Biomolecular Systems, London, UK | 4-tube strip | MIC-tubes |
| BioRad, Watford, UK | CFX Connect | N/A |
| BioRad, Watford, UK | CFX Opus | N/A |
| BioRad, Watford, UK | ddPCR probe supermix (-dUTP) | 186-3024 |
| BioRad, Watford, UK | CFX QX200 Droplet Generator | 186-4002 |
| BioRad, Watford, UK | CFX QX200 Droplet Reader | 186-4003 |
| BioRad, Watford, UK | Droplet generation oil for probes | 186-3005 |
| BioRad, Watford, UK | PX1 PCR plate sealer | 181-4000 |
| BioRad, Watford, UK | qPCR heat seal | 181-4030 |
| BioRad, Watford, UK | ddPCR heat seal | 181-4040 |
| BioRad, Watford, UK | Skirted 96-well plates | HSP9645 |
| BioRad, Watford, UK | CFX Maestro Software V. 2.3 | 12013758 |
| BioRad, Watford, UK | QX Manager Software V. 2.1. | N/A |
| Biosearch, Petaluma, CA, USA | EpiScript RNase H- Reverse Transcriptase | ERT12925K-ENZ |
| Cole Palmer, St., Neots, UK | Techne Prime Pro 48 cycler | WZ-93945-14 |
| Cole Palmer, St., Neots, UK | Techne plate seal | MB0481 |
| Cole Palmer, St., Neots, UK | Techne 48 well plates | MB0482 |
| IDT, Leuven, Belgium | PrimeTime Master Mix | 1055772 |
| Merck, Gillingham, UK | KAPA | KM4701 |
| NEB, Hitchin, UK | LyoPrime Luna Probe One-Step RT-qPCR Mix | L4001SVIAL |
| PCRBio, London, UK | qPCRBIO Probe Blue Mix | PB20.27-05 |
| PCRBio, London, UK | UltraScript 2 | PB30.33 |
| PCRBio, London, UK | UltraScript | PB30.12 |
| PCRBio, London, UK | Clara Probe 1-Step Mix | PB25.83-01 |
| Pentabase AS, Odense, DK | Pentabase primers | N/A |
| Premier Biosoft, San, Francisco, CA, USA | Beacon Designer 8.21 | N/A |
| Promega, Southampton, UK | GoTaq Probe qPCR master mix | A610A |
| Promega, Southampton, UK | GoTaq DNA polymerase | M7845 |
| Qiagen, Manchester, UK | RNeasy Mini Kit | 74104 |
| Qiagen, Manchester, UK | RNeasy Lipid Tissue Mini Kit | 74804 |
| Quanta, Beverly, MA, USA | PerfeCta qPCR Toughmix | 84196 |
| Quanta, Beverly, MA, USA | PerfeCta Multiplex qPCR Toughmix | 84263 |
| Sigma, Haverhill, Aldrich | Primers and probes | |
| Takara, Saint-Germain-en-Laye, F | Ex Taq probe premix | RR390 |
| Takara, Saint-Germain-en-Laye, F | ExTaq DNA polymerase | RR001A |
| ThermoFisher, Waltham, MA, USA | Nanodrop spectrophotometer | N2000 |
| ThermoFisher, Waltham, MA, USA | Nuclease-free water | AM9922 |
| ThermoFisher, Waltham, MA, USA | SuperScript IV | 18091200 |
| Qiagen, Manchester, UK | QiaQuant 96 | 9003000 |
| Qiagen, Manchester, UK | qPCR skirted plates | 209002 |
| Zymo, Research, Irvine,, CA, USA | Quick-RNA Miniprep Plus Kit | D7005 |
| 2× Buffer | B19 | B25 | B27 | B47 | B50 |
|---|---|---|---|---|---|
| Tris pH 8.8 (mM) | 20 | 20 | 20 | 20 | 20 |
| KCl (mM) | 100 | 100 | 100 | 100 | 100 |
| MgCl2 (mM) | 10 | 10 | 10 | 10 | 10 |
| 1,2 Propanediol (M) | 0.5 | 0.5 | 0.4 | 1.0 | 1.0 |
| 1,3 Propanediol (M) | 0.4 | 0.4 | 0.4 | - | - |
| Ethylene Glycol (M) | 0.6 | 0.6 | 0.6 | - | 0.16 |
| Trehalose (M) | 0.3 | 0.3 | 0.2 | 0.2 | 0.2 |
| BSA (mg/mL) | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 |
| dNTP (M) | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 |
| Formamide (%) | 0.5 | - | - | - | - |
| Target | Accession No | Primers | Sequence (5′-3′) | Tm (°C) | Amplicon |
|---|---|---|---|---|---|
| SARS-CoV-2 | NC_045512.2 | CoV-EF | GTGGTATTCTTGCTAGTTACACTAGCCATCC | 72.1 | 84bp |
| CoV-ER | AAGACTCACGTTAACAATATTGCAGCAGTAC | 71.2 | |||
| PB-F | TCTTGCTAGTTACACTAGCCATCC | 68.4 | 72bp | ||
| PB-R | TCACGTTAACAATATTGCAGCAGTAC | 68.2 | |||
| TNFα induced protein | NM_007115 | TSG-6F | TCGCAACTTACAAGCAGCTA | 65.8 | 85bp |
| TSG-6R | CCAACTCTGCCCTTAGCC | 66.1 | |||
| Hepatocyte growth factor 1 | NM_0006501 | HGF-1F | TCACAAGCAATCCAGAGGTAC | 65.9 | 76bp |
| HGF-1R | TTGCAGGTCATGCATTCAAC | 65.5 | |||
| GAPDH | NM_002046 | GAPDH-F | AGCCACATCGCTCAGACA | 67.4 | 75bp |
| GAPDH-R | TGACCAGGCGCCCAATAC | 68.5 | |||
| CDKN1A | NM_000389 | CDK-F | CTGGAGACTCTCAGGGTCGAA | 68.9 | 98bp |
| CDK-R | GGATTAGGGCTTCCTCTTGGA | 67.3 | |||
| Staphylococcus aureus | OR365499.1 | SA-F | GCGGTGAAATGCGCAGAGATATGGA | 73.3 | 77bp |
| SA-R | GCACATCAGCGTCAGTTACAGACCA | 72.9 | |||
| Candida auris | OQ581784.1 | CA-F | AACGGATCTCTTGGTTCTCGCATCG | 72.7 | 70bp |
| CA-R | CGTCTGCAAGTCATACTACGTATCGCAT | 72.2 | |||
| Acanthamoeba castellanii | KT185626.1 | Aca-F | GTCGATTGAACCTTACCATTTAGAGGAAGG | 70.8 | 74bp |
| Aca-R | GATCCCTCCGCAGGTTCACCTAC | 72.6 | |||
| Aspergillus fumigatus | OR415310.1 | Asp-F | TTCTTGGATTTGCTGAAGACTAACTACTGCG | 70.9 | 85bp |
| Asp-R | CGATCCCCTAACTTTCGTTCCCTGAT | 70.3 | |||
| Probes | Sequence (5′-3′) | Tm (°C) | Type | ||
| CoV-E-Pr | FAM-cacAcaAtcGaaGcgCagTaag-Q | 74.9 | LNA | ||
| PB-Pr | FAM-CACACAATCGAAGCGCAGTAAGGAT-Q | 72.5 | Pentabase | ||
| TSG-6-Pr | FAM-tccAtcCagCagCacaga-Q | 77.2 | LNA | ||
| HGF-1-Pr | HEX-cgaAgtCtgTgaCattcct-Q | 70.9 | LNA | ||
| GAPDH-Pr | FAM-tccGttcGacTccGacct-Q | 75 | LNA | ||
| CDK-Pr | FAM-atgCtgGtcTgcCgcc -Q | 77.4 | LNA | ||
| SA-Pr | HEX-acaCcaGtgGcgAagGcga-Q | 83.9 | LNA | ||
| CA-Pr | FAM-tcgCtgCgttCttCatCgat-Q | 76.7 | LNA | ||
| Aca-Pr | FAM-aagTcgTaaCaaGgtCtccg | 75.1 | LNA | ||
| Asp-Pr | FAM-acAtcCttGgcGaaTgcTttc-Q | 74.2 | LNA |
| qPCR Protocol | Reverse Transcription | Denaturation or Activation | Cycling | |||||
|---|---|---|---|---|---|---|---|---|
| Denaturation | Polymerisation | |||||||
| Temp (°C) | Time (min) | Temp (°C) | Time (s) | Temp (°C) | Time (s) | Temp (°C) | Time (s) | |
| P1 | N/A | N/A | 90 | 15 | 90 | 1 | 65 | 1 |
| P2 | N/A | N/A | 85 | 15 | 79-85 | 1 | 70 | 1 |
| P3 | N/A | N/A | 85 | 15 | 85 | 1 | 67–72 | 1 |
| P4 | N/A | N/A | 80 | 1 | 80 | 1 | 70 | 1 |
| P5 | N/A | N/A | 80 | 15 | 80 | 1 | 71 | 1 |
| P6 | N/A | N/A | 79 | 15 | 79 | 1 | 70 | 1 |
| P7 | N/A | N/A | 79 | 15 | 79 | 1 | 71 | 1 |
| P8 | N/A | N/A | 95 | 300 | 95 | 5 | 65 | 10 |
| P9 | N/A | N/A | 80 | 15 | 80 | 5 | 69 | 3 |
| P10 | N/A | N/A | 85 | 15 | 79–85 | 1 | 71 | 1 |
| P11 | N/A | N/A | 80 | 1 | 80 | 1 | 72 | 1 |
| P12 | N/A | N/A | 90 | 1 | 80–90 | 1 | 71 | 1 |
| P13 | N/A | N/A | 90 | 1 | 90 | 1 | 67–72 | 1 |
| P14 | N/A | N/A | 82 | 15 | 82 | 1 | 71 | 1 |
| P15 | N/A | N/A | 95 | 120 | 90 | 2 | 65 | 1 |
| RT-qPCR Protocol | ||||||||
| R1 | 50 | 5 | 95 | 120 | 90 | 2 | 65 | 1 |
| R2 | 50 | 5 | 95 | 60 | 95 | 1 | 60 | 1 |
| R3 | Room Temp | 5 | 95 | 60 | 95 | 1 | 60 | 1 |
| R4 | Room Temp | 5 | 90 | 15 | 90 | 1 | 60 | 1 |
| R5 | Room Temp | 5 | 81 | 15 | 81 | 1 | 70 | 1 |
| R6 | Room Temp | 5 | 80 | 15 | 80 | 1 | 70 | 1 |
| R7 | Room Temp | 5 | 80 | 15 | 80 | 1 | 71 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustin, S.A.; Kirvell, S.; Nolan, T.; Shipley, G.L. FlashPCR: Revolutionising qPCR by Accelerating Amplification through Low ∆T Protocols. Int. J. Mol. Sci. 2024, 25, 2773. https://doi.org/10.3390/ijms25052773
Bustin SA, Kirvell S, Nolan T, Shipley GL. FlashPCR: Revolutionising qPCR by Accelerating Amplification through Low ∆T Protocols. International Journal of Molecular Sciences. 2024; 25(5):2773. https://doi.org/10.3390/ijms25052773
Chicago/Turabian StyleBustin, Stephen A., Sara Kirvell, Tania Nolan, and Gregory L. Shipley. 2024. "FlashPCR: Revolutionising qPCR by Accelerating Amplification through Low ∆T Protocols" International Journal of Molecular Sciences 25, no. 5: 2773. https://doi.org/10.3390/ijms25052773
APA StyleBustin, S. A., Kirvell, S., Nolan, T., & Shipley, G. L. (2024). FlashPCR: Revolutionising qPCR by Accelerating Amplification through Low ∆T Protocols. International Journal of Molecular Sciences, 25(5), 2773. https://doi.org/10.3390/ijms25052773