
Mitochondrial Dysfunction and Metabolic Reprogramming in Obesity and Asthma
Abstract
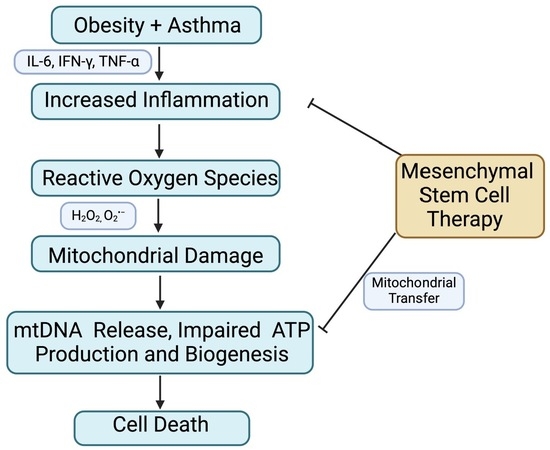
:
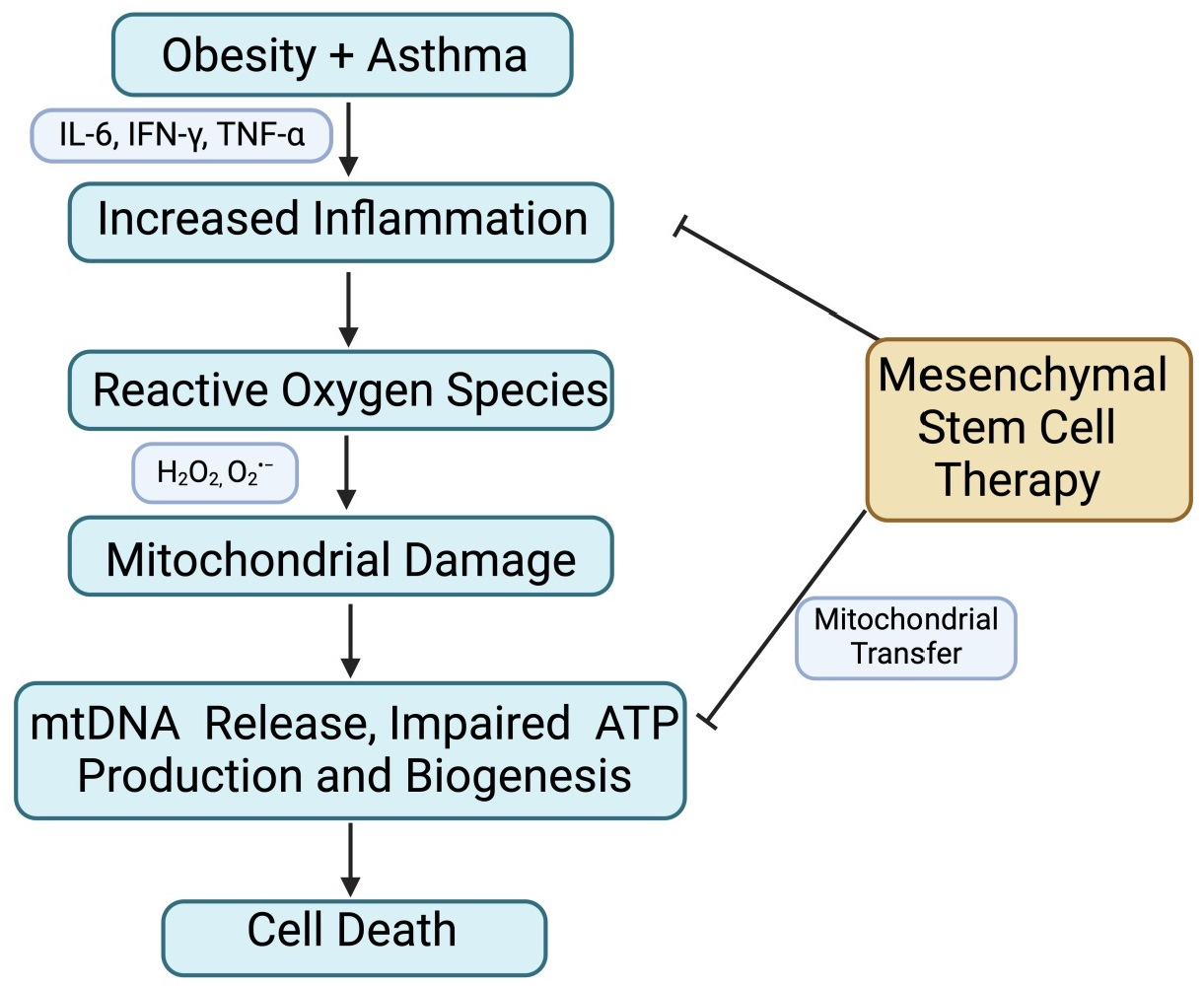{kind=link}
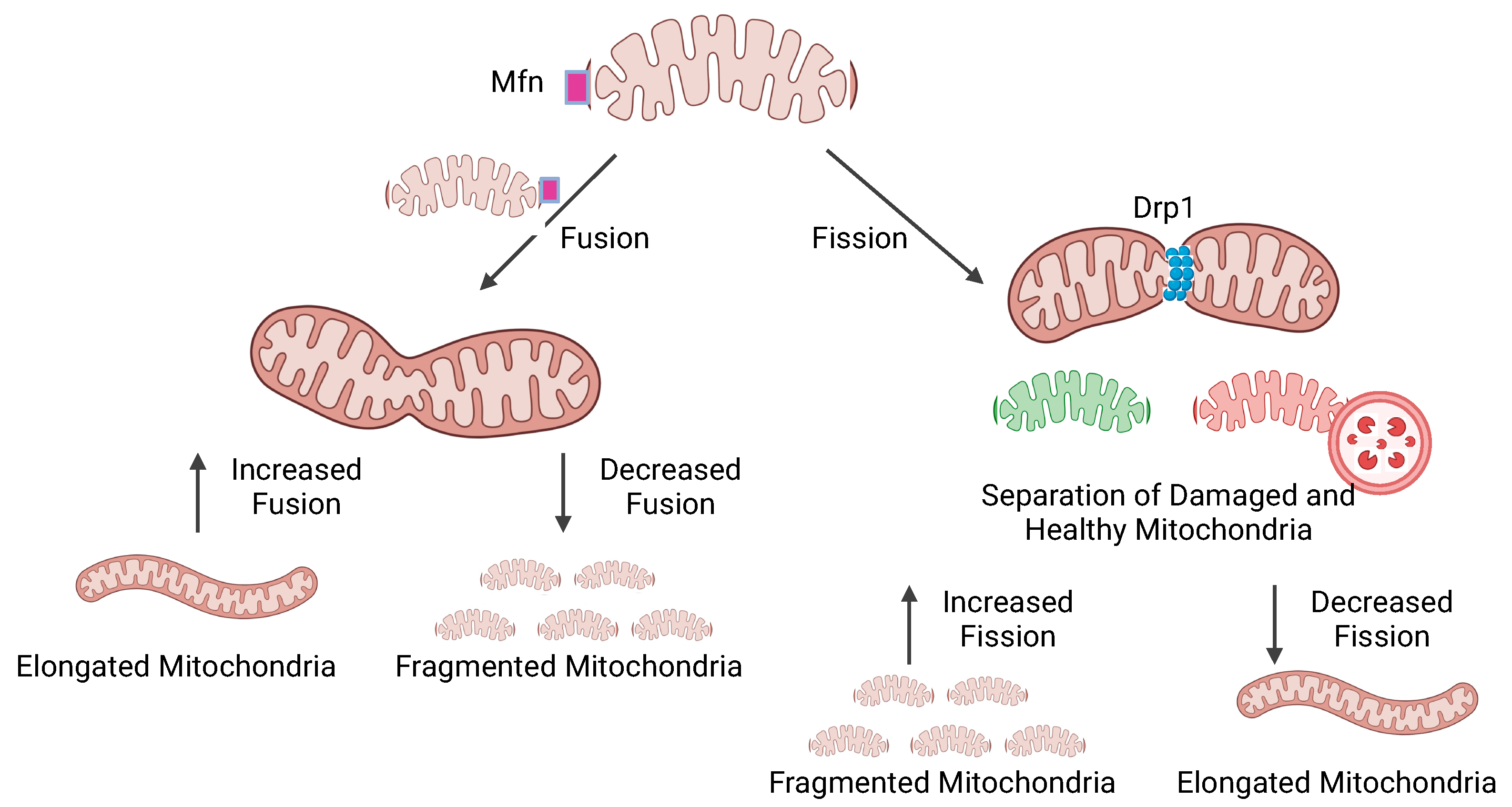{kind=link}
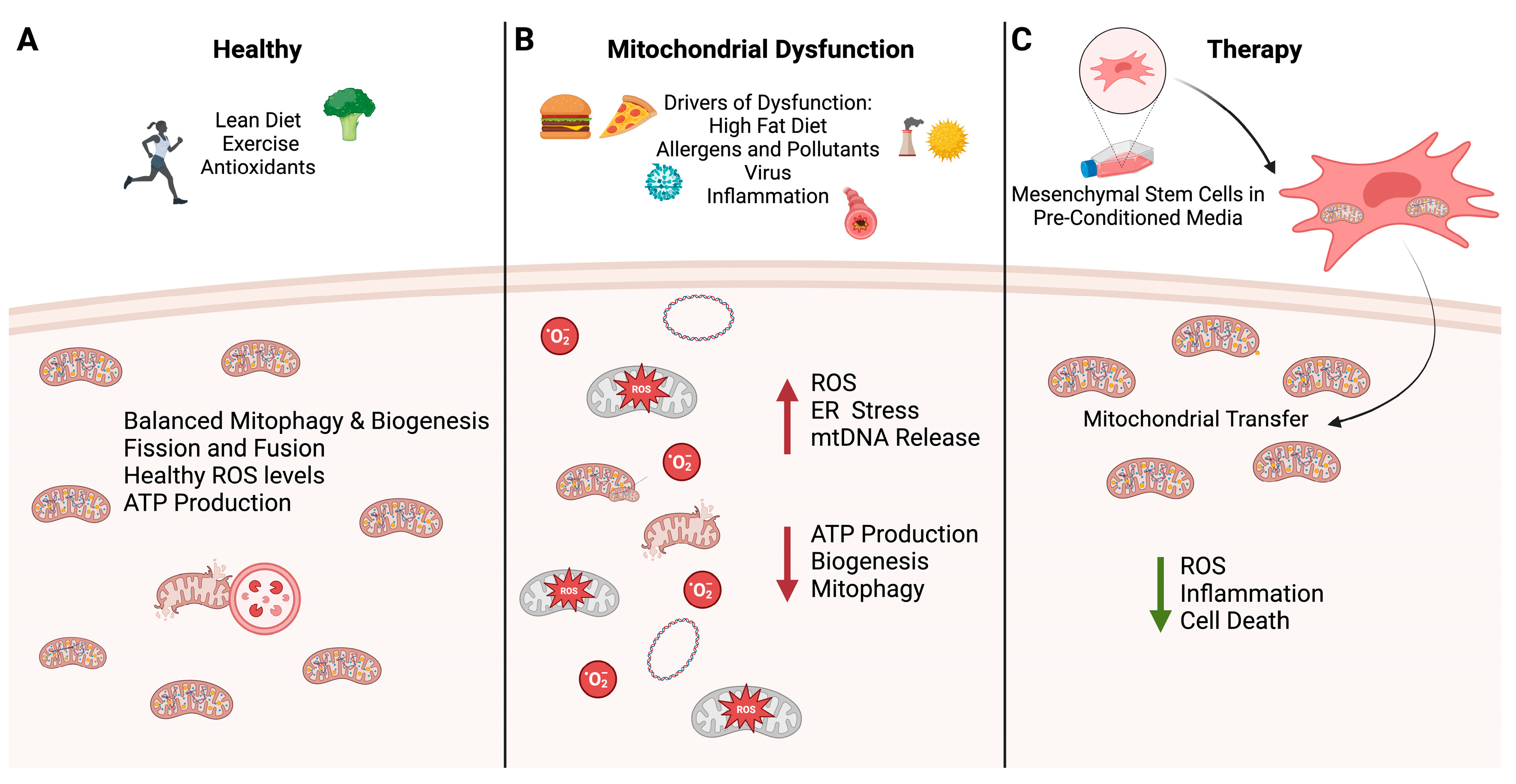{kind=link}
1. Introduction
2. Role of the Mitochondria in Health
3. Mitochondrial Dysfunction and Metabolic Reprogramming
4. Common Forms of Mitochondrial Dysfunction and Metabolic Reprogramming
5. Mitochondrial Dysfunction in Obesity and Asthma
6. Mitochondrial Dysfunction in Other Diseases
7. Mechanisms Driving Mitochondrial Dysfunction
8. Therapeutic Approaches to Restore Mitochondrial Functions in Diseases
9. Cell-Based Therapeutic Approach: Mesenchymal Stem Cells
10. Final Remarks
- (1)
- What are the unique features or biomarkers of mitochondrial dysfunction and metabolic reprogramming in obese asthma as compared to lean asthma and healthy subjects?
- (2)
- Do mitochondrial dysfunction and metabolic reprogramming profiles predict the clinical and immunological phenotypes or endotypes such as pulmonary function and inflammation?
- (3)
- How may research findings from profiling mitochondrial dysfunction and metabolic reprogramming guide precision medicine in obese asthma?
- (4)
- Can we develop or refine mitochondria-targeted therapies to restore mitochondrial and metabolic homeostasis?
- (5)
- Are physical and dietary approaches effective in restoring mitochondrial function and metabolism in patients with obese asthma?
- (1)
- Perform cross-sectional and longitudinal studies in large cohorts of subjects with obese asthma and controls to obtain biological and metabolic data reflecting mitochondrial dysfunction and metabolic reprogramming. Research data will be analyzed in the context of clinical and immunological phenotypes or endotypes.
- (2)
- Develop and validate blood or airway biomarkers using minimally invasive approaches such as nasal brushing or lavage to indicate the levels of mitochondrial dysfunction and metabolic reprogramming in obese asthma.
- (3)
- Develop patient-specific therapies to restore mitochondrial homeostasis and improve clinical outcomes. This precision medicine approach needs more robust cutting-edge research to define how an individual subject with obese asthma presents a unique profile of mitochondrial dysfunction and metabolic reprogramming.
- (4)
- It is unlikely that a single mitochondria-target therapy will be effective to improve airway function in subjects with obese asthma. A combinational therapy targeting several pathways such as ROS and glycolysis may be necessary to maximize the therapeutic efficiency. Additionally, the interactions of mitochondria-target therapy and common asthma medications such as bronchodilators and corticosteroids should be considered to increase the therapeutic potency, while reducing any potential side effects.
- (5)
- Any mitochondria-target therapy (e.g., use of MSCs) should be combined with increased physical activity and an improved diet to further reduce pathobiological effects of mitochondrial dysfunction and metabolic reprogramming in obese asthma.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nunes, C.; Pereira, A.M.; Morais-Almeida, M. Asthma costs and social impact. Asthma Res. Pract. 2017, 3, 1. [Google Scholar] [CrossRef]
- Fahy, J.V. Type 2 inflammation in asthma--present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef]
- Taylor, B.; Mannino, D.; Brown, C.; Crocker, D.; Twum-Baah, N.; Holguin, F. Body mass index and asthma severity in the National Asthma Survey. Thorax 2008, 63, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.; Denton, E. Asthma in Children and Adults-What Are the Differences and What Can They Tell us About Asthma? Front. Pediatr. 2019, 7, 256. [Google Scholar] [CrossRef]
- Winnica, D.; Corey, C.; Mullett, S.; Reynolds, M.; Hill, G.; Wendell, S.; Que, L.; Holguin, F.; Shiva, S. Bioenergetic Differences in the Airway Epithelium of Lean Versus Obese Asthmatics Are Driven by Nitric Oxide and Reflected in Circulating Platelets. Antioxid. Redox Signal 2019, 31, 673–686. [Google Scholar] [CrossRef]
- Bseikri, M.; McCann, J.C.; Lal, A.; Fong, E.; Graves, K.; Goldrich, A.; Block, D.; Gildengoren, G.L.; Mietus-Snyder, M.; Shigenaga, M.; et al. A novel nutritional intervention improves lung function in overweight/obese adolescents with poorly controlled asthma: The Supplemental Nutrition in Asthma Control (SNAC) pilot study. FASEB J. 2018, 32, 6643–6654. [Google Scholar] [CrossRef] [PubMed]
- Kasteleyn, M.J.; Bonten, T.N.; de Mutsert, R.; Thijs, W.; Hiemstra, P.S.; le Cessie, S.; Rosendaal, F.R.; Chavannes, N.H.; Taube, C. Pulmonary function, exhaled nitric oxide and symptoms in asthma patients with obesity: A cross-sectional study. Respir. Res. 2017, 18, 205. [Google Scholar] [CrossRef]
- Sideleva, O.; Suratt, B.T.; Black, K.E.; Tharp, W.G.; Pratley, R.E.; Forgione, P.; Dienz, O.; Irvin, C.G.; Dixon, A.E. Obesity and asthma: An inflammatory disease of adipose tissue not the airway. Am. J. Respir. Crit. Care Med. 2012, 186, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, J.; Zhang, L.; Liu, Y.; Chen, G.P.; Zhang, H.P.; Wang, L.; Kang, Y.; Wood, L.G.; Wang, G. Systemic inflammation mediates the detrimental effects of obesity on asthma control. Allergy Asthma Proc. 2018, 39, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef]
- Aguilera-Aguirre, L.; Bacsi, A.; Saavedra-Molina, A.; Kurosky, A.; Sur, S.; Boldogh, I. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 2009, 183, 5379–5387. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Asthma: Implications for Mitochondria-Targeted Antioxidant Therapeutics. Pharmaceuticals 2011, 4, 429–456. [Google Scholar] [CrossRef]
- Xu, W.; Ghosh, S.; Comhair, S.A.; Asosingh, K.; Janocha, A.J.; Mavrakis, D.A.; Bennett, C.D.; Gruca, L.L.; Graham, B.B.; Queisser, K.A.; et al. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J. Clin. Investig. 2016, 126, 2465–2481. [Google Scholar] [CrossRef]
- Pang, Y.; Zhang, C.; Gao, J. Macrophages as Emerging Key Players in Mitochondrial Transfers. Front. Cell Dev. Biol. 2021, 9, 747377. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best. Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef]
- Elesela, S.; Lukacs, N.W. Role of Mitochondria in Viral Infections. Life 2021, 11, 232. [Google Scholar] [CrossRef]
- Faas, M.M.; de Vos, P. Mitochondrial function in immune cells in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165845. [Google Scholar] [CrossRef]
- Hanada, Y.; Ishihara, N.; Wang, L.; Otera, H.; Ishihara, T.; Koshiba, T.; Mihara, K.; Ogawa, Y.; Nomura, M. MAVS is energized by Mff which senses mitochondrial metabolism via AMPK for acute antiviral immunity. Nat. Commun. 2020, 11, 5711. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; Gonzalez-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vazquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41, 491–517. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Bhavsar, P.K.; Mumby, S.; Xu, B.; Hui, C.K.M.; Chung, K.F.; Adcock, I.M. Role of Metabolic Reprogramming in Pulmonary Innate Immunity and Its Impact on Lung Diseases. J. Innate Immun. 2020, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Dennery, P.A.; Yao, H. Metabolic reprogramming in the pathogenesis of chronic lung diseases, including BPD, COPD, and pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L544–L554. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Kim, K.; Esteves, P.; Trian, T.; Barnes, P.J. Mitochondrial dysfunction in lung ageing and disease. Eur. Respir. Rev. 2020, 29, 200165. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 5, e126915. [Google Scholar] [CrossRef]
- Newell, C.; Sabouny, R.; Hittel, D.S.; Shutt, T.E.; Khan, A.; Klein, M.S.; Shearer, J. Mesenchymal Stem Cells Shift Mitochondrial Dynamics and Enhance Oxidative Phosphorylation in Recipient Cells. Front. Physiol. 2018, 9, 1572. [Google Scholar] [CrossRef]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Stepien, K.M.; Heaton, R.; Rankin, S.; Murphy, A.; Bentley, J.; Sexton, D.; Hargreaves, I.P. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J. Clin. Med. 2017, 6, 71. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Osko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczynska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Schuliga, M.; Kanwal, A.; Read, J.; Blokland, K.E.C.; Burgess, J.K.; Prele, C.M.; Mutsaers, S.E.; Grainge, C.; Thomson, C.; James, A.; et al. A cGAS-dependent response links DNA damage and senescence in alveolar epithelial cells: A potential drug target in IPF. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L859–L871. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Lurbet, M.F.; Rojano, B.; Whittaker Brown, S.A.; Busse, P.; Holguin, F.; Federman, A.D.; Wisnivesky, J.P. Obesity Trends among Asthma Patients in the United States: A Population-based Study. Ann. Glob. Health 2019, 85, 10. [Google Scholar] [CrossRef] [PubMed]
- Akinbami, L.J.; Fryar, C.D. Current Asthma Prevalence by Weight Status Among Adults: United States, 2001–2014. NCHS Data Brief 2016, 239, 1–8. [Google Scholar]
- Ellulu, M.S.; Patimah, I.; Khaza'ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Xu, S.; Karmacharya, N.; Cao, G.; Guo, C.; Gow, A.; Panettieri, R.A., Jr.; Jude, J.A. Obesity elicits a unique metabolomic signature in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2022, 323, L297–L307. [Google Scholar] [CrossRef]
- Holguin, F.; Comhair, S.A.; Hazen, S.L.; Powers, R.W.; Khatri, S.S.; Bleecker, E.R.; Busse, W.W.; Calhoun, W.J.; Castro, M.; Fitzpatrick, A.M.; et al. An association between L-arginine/asymmetric dimethyl arginine balance, obesity, and the age of asthma onset phenotype. Am. J. Respir. Crit. Care Med. 2013, 187, 153–159. [Google Scholar] [CrossRef]
- Maniscalco, M.; Paris, D.; Melck, D.J.; D'Amato, M.; Zedda, A.; Sofia, M.; Stellato, C.; Motta, A. Coexistence of obesity and asthma determines a distinct respiratory metabolic phenotype. J. Allergy Clin. Immunol. 2017, 139, 1536–1547. [Google Scholar] [CrossRef]
- Carpagnano, G.E.; Lacedonia, D.; Carone, M.; Soccio, P.; Cotugno, G.; Palmiotti, G.A.; Scioscia, G.; Foschino Barbaro, M.P. Study of mitochondrial DNA alteration in the exhaled breath condensate of patients affected by obstructive lung diseases. J. Breath. Res. 2016, 10, 026005. [Google Scholar] [CrossRef] [PubMed]
- Carpagnano, G.E.; Scioscia, G.; Lacedonia, D.; Soccio, P.; Quarato, C.M.I.; Cotugno, G.; Palumbo, M.G.; Foschino Barbaro, M.P. Searching for Inflammatory and Oxidative Stress Markers Capable of Clustering Severe Asthma. Arch. Bronconeumol. 2021, 57, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Prakash, Y.S. Obesity, metabolic syndrome, and airway disease: A bioenergetic problem? Immunol. Allergy Clin. North. Am. 2014, 34, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Hospital, C.; Tete, A.; Brial, F.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Kim, M.J.; Blanc, E.B.; Coumoul, X.; Bortoli, S. Mitochondrial Dysfunction as a Hallmark of Environmental Injury. Cells 2021, 11, 110. [Google Scholar] [CrossRef]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef]
- Fernandez-Sanchez, A.; Madrigal-Santillan, E.; Bautista, M.; Esquivel-Soto, J.; Morales-Gonzalez, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sanchez-Rivera, G.; Valadez-Vega, C.; Morales-Gonzalez, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Jayashankar, V.; Selwan, E.; Hancock, S.E.; Verlande, A.; Goodson, M.O.; Eckenstein, K.H.; Milinkeviciute, G.; Hoover, B.M.; Chen, B.; Fleischman, A.G.; et al. Drug-like sphingolipid SH-BC-893 opposes ceramide-induced mitochondrial fission and corrects diet-induced obesity. EMBO Mol. Med. 2021, 13, e13086. [Google Scholar] [CrossRef]
- Wang, Y.; McLean, A.S. The Role of Mitochondria in the Immune Response in Critical Illness. Crit. Care 2022, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Lacedonia, D.; Carpagnano, G.E.; Crisetti, E.; Cotugno, G.; Palladino, G.P.; Patricelli, G.; Sabato, R.; Foschino Barbaro, M.P. Mitochondrial DNA alteration in obstructive sleep apnea. Respir. Res. 2015, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal 2014, 20, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson's Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Cai, S.; Zhao, M.; Zhou, B.; Yoshii, A.; Bugg, D.; Villet, O.; Sahu, A.; Olson, G.S.; Davis, J.; Tian, R. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 2023, 133, e159498. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Xu, Y.; Xue, D.; Bankhead, A., 3rd; Neamati, N. Why All the Fuss about Oxidative Phosphorylation (OXPHOS)? J. Med. Chem. 2020, 63, 14276–14307. [Google Scholar] [CrossRef] [PubMed]
- Bhatraju, N.K.; Agrawal, A. Mitochondrial Dysfunction Linking Obesity and Asthma. Ann. Am. Thorac. Soc. 2017, 14, S368–S373. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, L.; Weber, S.; Elgizouli, M.; Stoehlker, A.S.; Geist, I.; Peter, H.H.; Vach, W.; Nieters, A. Obesity and risk of respiratory tract infections: Results of an infection-diary based cohort study. BMC Public. Health 2018, 18, 271. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Henderson, R.J.; Holbrook, J.T.; Que, L.G.; Mathews, A.M.; Wise, R.A.; Dixon, A.E.; Peters, S.P.; Rogers, L.; Smith, L.J.; et al. Does Obesity Increase Respiratory Tract Infections in Patients with Asthma? J. Allergy Clin. Immunol. Pract. 2019, 7, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Vanessa, M.; Powell, H.; Gibson, P. The role of maternal obesity in susceptibility to respiratory viral infection and exacerbation rate in pregnant women with asthma. Eur. Respir. J. 2014, 44, 224. [Google Scholar]
- Forno, E.; Litonjua, A.A. Pollution, Obesity, Vitamin D, or Why Is Asthma So Complicated-and Interesting. J. Allergy Clin. Immunol. Pract. 2019, 7, 1823–1824. [Google Scholar] [CrossRef] [PubMed]
- Dimasuay, K.G.; Berg, B.; Schaunaman, N.; Holguin, F.; Winnica, D.; Chu, H.W. High-fat diet and palmitic acid amplify airway type 2 inflammation. Front. Allergy 2023, 4, 1193480. [Google Scholar] [CrossRef]
- Mick, E.; Titov, D.V.; Skinner, O.S.; Sharma, R.; Jourdain, A.A.; Mootha, V.K. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife 2020, 9, e49178. [Google Scholar] [CrossRef]
- Spier, A.; Stavru, F.; Cossart, P. Interaction between Intracellular Bacterial Pathogens and Host Cell Mitochondria. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef]
- Chew, S.; Kolosowska, N.; Saveleva, L.; Malm, T.; Kanninen, K.M. Impairment of mitochondrial function by particulate matter: Implications for the brain. Neurochem. Int. 2020, 135, 104694. [Google Scholar] [CrossRef]
- Chew, S.; Lampinen, R.; Saveleva, L.; Korhonen, P.; Mikhailov, N.; Grubman, A.; Polo, J.M.; Wilson, T.; Komppula, M.; Ronkko, T.; et al. Urban air particulate matter induces mitochondrial dysfunction in human olfactory mucosal cells. Part. Fibre Toxicol. 2020, 17, 18. [Google Scholar] [CrossRef]
- Chavakis, T. Immunometabolism: Where Immunology and Metabolism Meet. J. Innate Immun. 2022, 14, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, I.W.; Curto, A.; Lopez-Vicario, C.; Casulleras, M.; Duran-Guell, M.; Flores-Costa, R.; Colsch, B.; Aguilar, F.; Aransay, A.M.; Lozano, J.J.; et al. Mitochondrial dysfunction governs immunometabolism in leukocytes of patients with acute-on-chronic liver failure. J. Hepatol. 2022, 76, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Eneli, I.U.; Skybo, T.; Camargo, C.A., Jr. Weight loss and asthma: A systematic review. Thorax 2008, 63, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Juel, C.T.; Ali, Z.; Nilas, L.; Ulrik, C.S. Asthma and obesity: Does weight loss improve asthma control? a systematic review. J. Asthma Allergy 2012, 5, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Bantula, M.; Tubita, V.; Roca-Ferrer, J.; Mullol, J.; Valero, A.; Bobolea, I.; Pascal, M.; de Hollanda, A.; Vidal, J.; Picado, C.; et al. Weight Loss and Vitamin D Improve Hyporesponsiveness to Corticosteroids in Obese Asthma. J. Investig. Allergol. Clin. Immunol. 2023, 33, 464–473. [Google Scholar] [CrossRef]
- Seo, M.; Kim, H.; Noh, H.; Jeon, J.S.; Byun, D.W.; Kim, S.H.; Kim, H.J.; Suh, K.; Park, H.K.; Kwon, S.H. Effect of bariatric surgery on circulating and urinary mitochondrial DNA copy numbers in obesity with or without diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001372. [Google Scholar] [CrossRef]
- Qian, L.; Mehrabi Nasab, E.; Athari, S.M.; Athari, S.S. Mitochondria signaling pathways in allergic asthma. J. Investig. Med. 2022, 70, 863–882. [Google Scholar] [CrossRef]
- Chong, L.; Li, H.; Zhu, L.; Yu, G. Regulatory effect of mitoQ on the mtROS-NLRP3 inflammasome pathway in leptin-pretreated BEAS-2 cells. Exp. Ther. Med. 2021, 21, 466. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Peault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef]
- Yao, P.; Zhou, L.; Zhu, L.; Zhou, B.; Yu, Q. Mesenchymal Stem Cells: A Potential Therapeutic Strategy for Neurodegenerative Diseases. Eur. Neurol. 2020, 83, 235–241. [Google Scholar] [CrossRef]
- de Castro, L.L.; Xisto, D.G.; Kitoko, J.Z.; Cruz, F.F.; Olsen, P.C.; Redondo, P.A.G.; Ferreira, T.P.T.; Weiss, D.J.; Martins, M.A.; Morales, M.M.; et al. Human adipose tissue mesenchymal stromal cells and their extracellular vesicles act differentially on lung mechanics and inflammation in experimental allergic asthma. Stem Cell Res. Ther. 2017, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.B.; Zhang, H.Y.; Wang, C.; He, B.X.; Liu, X.Q.; Meng, X.C.; Peng, Y.Q.; Xu, Z.B.; Fan, X.L.; Wu, Z.J.; et al. Small extracellular vesicles derived from human mesenchymal stromal cells prevent group 2 innate lymphoid cell-dominant allergic airway inflammation through delivery of miR-146a-5p. J. Extracell. Vesicles 2020, 9, 1723260. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, Y.; Zeng, J.; Chang, N.; Cheng, Y.; Zhen, X.; Zhong, D.; Chen, R.; Ma, G.; Wang, Y. Mesenchymal Stem/Stromal Cells in Asthma Therapy: Mechanisms and Strategies for Enhancement. Cell Transplant. 2023, 32, 9636897231180128. [Google Scholar] [CrossRef]
- Pham, D.V.; Nguyen, T.K.; Park, P.H. Adipokines at the crossroads of obesity and mesenchymal stem cell therapy. Exp. Mol. Med. 2023, 55, 313–324. [Google Scholar] [CrossRef]
- Mohammadalipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 603292. [Google Scholar] [CrossRef]
- Kim, J.; Hematti, P. Mesenchymal stem cell-educated macrophages: A novel type of alternatively activated macrophages. Exp. Hematol. 2009, 37, 1445–1453. [Google Scholar] [CrossRef]
- Sagar, S.; Faizan, M.I.; Chaudhary, N.; Singh, V.; Singh, P.; Gheware, A.; Sharma, K.; Azmi, I.; Singh, V.P.; Kharya, G.; et al. Obesity impairs cardiolipin-dependent mitophagy and therapeutic intercellular mitochondrial transfer ability of mesenchymal stem cells. Cell Death Dis. 2023, 14, 324. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Wilen, C.B.; Moley, J.R.; Li, Y.; Zou, W.; Malvin, N.P.; Rowen, M.N.; Saunders, B.T.; Ma, H.; Mack, M.R.; et al. Intercellular Mitochondria Transfer to Macrophages Regulates White Adipose Tissue Homeostasis and Is Impaired in Obesity. Cell Metab. 2021, 33, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Eggenhofer, E.; Benseler, V.; Kroemer, A.; Popp, F.C.; Geissler, E.K.; Schlitt, H.J.; Baan, C.C.; Dahlke, M.H.; Hoogduijn, M.J. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front. Immunol. 2012, 3, 297. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, L. Preconditioning influences mesenchymal stem cell properties in vitro and in vivo. J. Cell Mol. Med. 2018, 22, 1428–1442. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartsoe, P.; Holguin, F.; Chu, H.W. Mitochondrial Dysfunction and Metabolic Reprogramming in Obesity and Asthma. Int. J. Mol. Sci. 2024, 25, 2944. https://doi.org/10.3390/ijms25052944
Hartsoe P, Holguin F, Chu HW. Mitochondrial Dysfunction and Metabolic Reprogramming in Obesity and Asthma. International Journal of Molecular Sciences. 2024; 25(5):2944. https://doi.org/10.3390/ijms25052944
Chicago/Turabian StyleHartsoe, Paige, Fernando Holguin, and Hong Wei Chu. 2024. "Mitochondrial Dysfunction and Metabolic Reprogramming in Obesity and Asthma" International Journal of Molecular Sciences 25, no. 5: 2944. https://doi.org/10.3390/ijms25052944
APA StyleHartsoe, P., Holguin, F., & Chu, H. W. (2024). Mitochondrial Dysfunction and Metabolic Reprogramming in Obesity and Asthma. International Journal of Molecular Sciences, 25(5), 2944. https://doi.org/10.3390/ijms25052944