

Regulation of Cell Cycle Progression through RB Phosphorylation by Nilotinib and AT-9283 in Human Melanoma A375P Cells
 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Predicting the Effect of BCR-ABL Kinase Inhibitors on Skin Cancer Cells
2.2. BCR-ABL Kinase Inhibitors Suppress Proliferation and Migration of Human Melanoma A375P Cells
2.3. AT-9283 and Nilotinib Can Control Expression of Cell Cycle-Associated Genes in A375P Cells
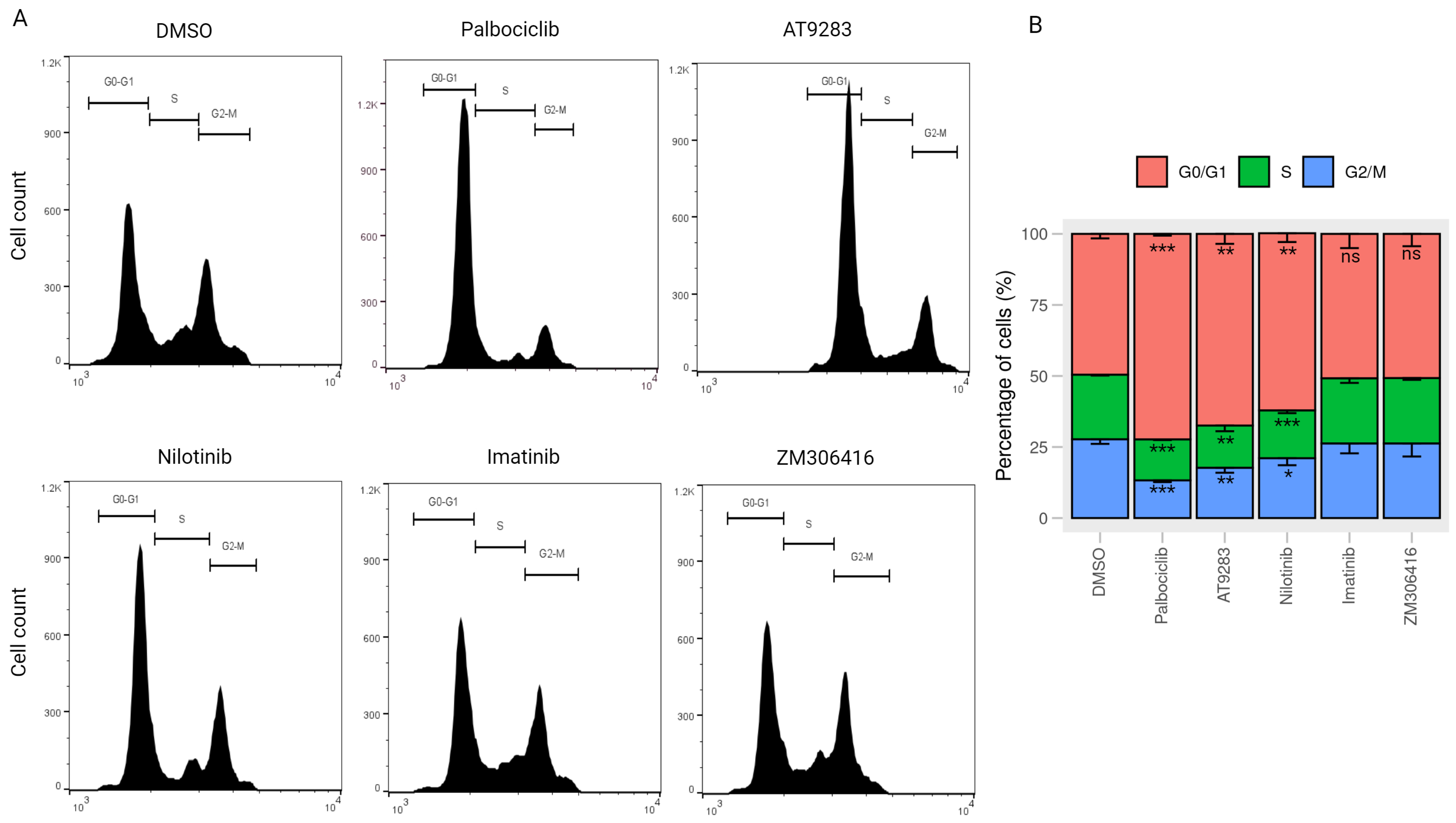
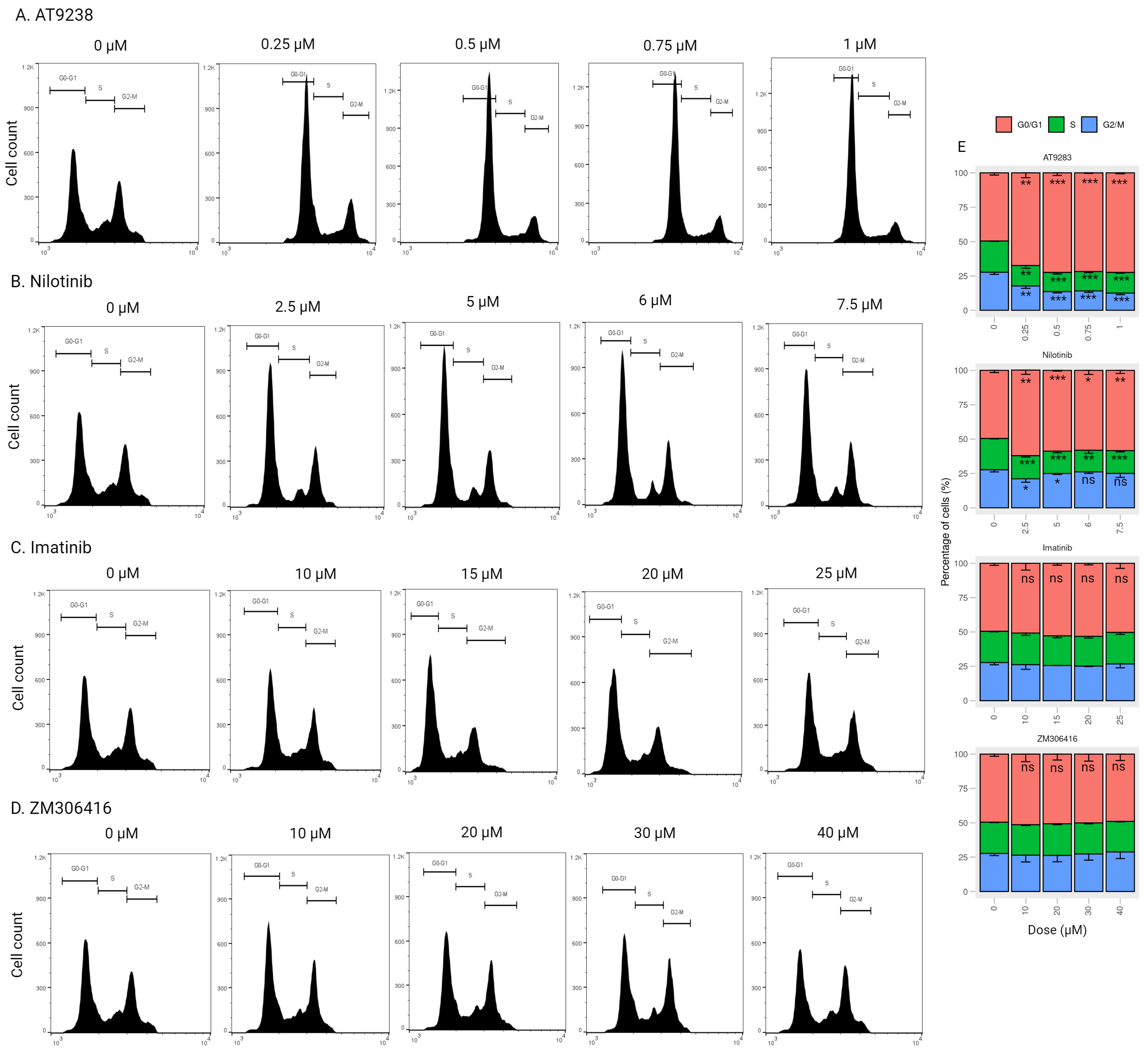
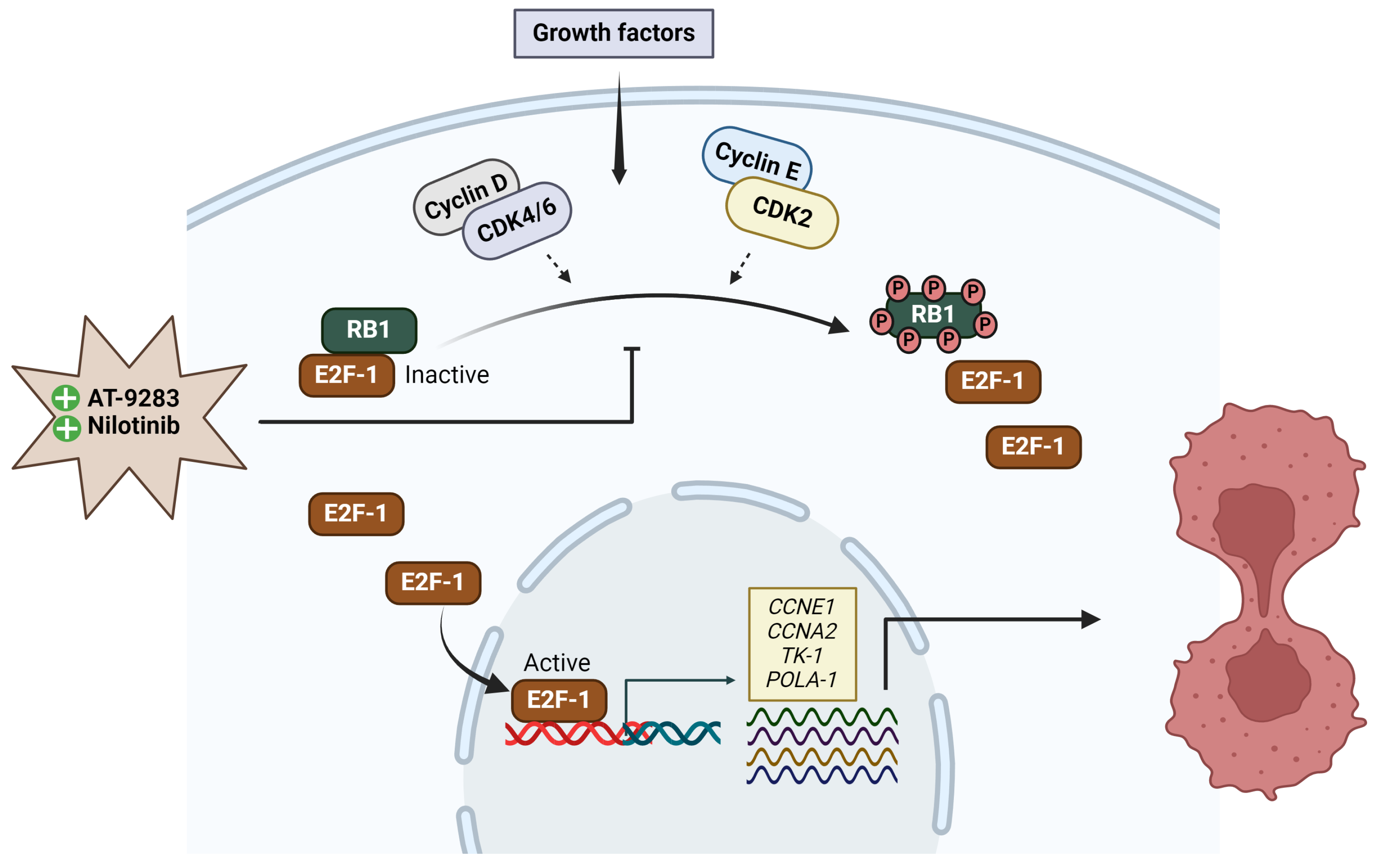
2.4. AT-9283 and Nilotinib Regulate the Cell Cycle in a RB/E2F-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Identification of Targets for Tyrosine Kinase Inhibitors in Proliferating Cancer Cells
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. BrdU Cell Proliferation Assay
4.6. Wound-Healing Assay
4.7. Cell Cycle Analysis by Flow Cytometry
4.8. Western Blot Analysis
4.9. RNA Extraction and RT-qPCR Analysis
4.10. Subcellular Fractionation
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pan, Z.; Zhang, X.; Yu, P.; Chen, X.; Lu, P.; Li, M.; Liu, X.; Li, Z.; Wei, F.; Wang, K.; et al. Cinobufagin Induces Cell Cycle Arrest at the G2/M Phase and Promotes Apoptosis in Malignant Melanoma Cells. Front. Oncol. 2019, 9, 853. [Google Scholar] [CrossRef] [PubMed]
- Velho, T.R. Metastatic melanoma—A review of current and future drugs. Drugs Context 2012, 2012, 212242. [Google Scholar] [CrossRef] [PubMed]
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer What Does the Future Hold. Dermatol. Pract. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Prieto-Granada, C.; Howe, N.; McCardle, T. Melanoma Pathology. In Melanoma; Oxford University Press: Oxford, UK, 2016; Volume 1012, pp. 10–30. [Google Scholar] [CrossRef]
- Kimbrough, C.W.; McMasters, K.M.; Davis, E.G. Principles of surgical treatment of malignant melanoma. Surg. Clin. North Am. 2014, 94, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Kalal, B.S.; Upadhya, D.; Pai, V.R. Chemotherapy Resistance Mechanisms in Advanced Skin Cancer. Oncol. Rev. 2017, 11, 19–25. [Google Scholar] [CrossRef]
- Janostiak, R.; Torres-Sanchez, A.; Posas, F.; de Nadal, E. Understanding Retinoblastoma Post-Translational Regulation for the Design of Targeted Cancer Therapies. Cancers 2022, 14, 1265. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Donjerkovic, D.; Scott, D.W. Regulation of the G1 phase of the mammalian cell cycle. Cell Res. 2000, 10, 1–16. [Google Scholar] [CrossRef]
- Rubin, S.M.; Sage, J.; Skotheim, J.M. Integrating Old and New Paradigms of G1/S Control. Mol. Cell 2020, 80, 183–192. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; De Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: P53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Goldman, J.M.; Melo, J.V. BCR-ABL in chronic myelogenous leukemia—How does it work? Acta Haematol. 2008, 119, 212–217. [Google Scholar] [CrossRef]
- Liang, X.; Hajivandi, M.; Veach, D.; Wisniewski, D.; Clarkson, B.; Resh, M.D.; Pope, R.M. Quantification of change in phosphorylation of BCR-ABL kinase and its substrates in response to Imatinib treatment in human chronic myelogenous leukemia cells. Proteomics 2006, 6, 4554–4564. [Google Scholar] [CrossRef] [PubMed]
- Mughal, A.; Aslam, H.M.; Khan, A.M.H.; Saleem, S.; Umah, R.; Saleem, M. Bcr-Abl tyrosine kinase inhibitors- current status. Infect. Agents Cancer 2013, 8, 23. [Google Scholar] [CrossRef]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Cortés, J.E.; Kantarjian, H. Practical management of toxicities associated with tyrosine kinase inhibitors in chronic myeloid leukemia. Clin. Lymphoma Myeloma 2008, 8 (Suppl. 3), S82–S88. [Google Scholar] [CrossRef]
- Nowell, C. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut 1962, 8, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Holtz, M.; Gupta, M.; Bhatia, R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood 2004, 103, 3167–3174. [Google Scholar] [CrossRef]
- Oetzel, C.; Jonuleit, T.; Götz, A.; Van Der Kuip, H.; Michels, H.; Duyster, J.; Hallek, M.; Aulitzky, W.E. The tyrosine kinase inhibitor CGP 57148 (ST1 571) induces apoptosis in BCR-ABL-positive cells by down-regulating BCL-X. Clin. Cancer Res. 2000, 6, 1958–1968. [Google Scholar]
- Deininger, M.W.; Goldman, J.M.; Lydon, N.; Melo, J.V. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood 1997, 90, 3691–3698. [Google Scholar] [CrossRef]
- Ahmed, M.; Kim, D.R. LINPS: A database for cancer-cell-specific perturbations of biological networks. Database 2021, 2021, 1–6. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J.; Abraham, D.; Norman, J. Platelet-derived growth factor signaling in mesenchymal cells. Front. Biosci. Landmark Ed. 2013, 18, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Abouantoun, T.J.; MacDonald, T.J. Imatinib blocks migration and invasion of medulloblastoma cells by concurrently inhibiting activation of platelet-derived growth factor receptor and transactivation of epidermal growth factor receptor. Mol. Cancer Ther. 2009, 8, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.S.; Xing, X.F.; Zeng, P.; Zhou, X. Determination of propranolol in human plasma by reversed phase high performance liquid chromatography. Acta Pharm. Sin. 1987, 22, 238–240. [Google Scholar]
- Wang, S.; Xie, Y.; Bao, A.; Li, J.; Ye, T.; Yang, C.; Yu, S. Nilotinib, a Discoidin domain receptor 1 (DDR1) inhibitor, induces apoptosis and inhibits migration in breast cancer. Neoplasma 2021, 68, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165715. [Google Scholar] [CrossRef] [PubMed]
- Ezhevsky, S.A.; Nagahara, H.; Vocero-Akbani, A.M.; Gius, D.R.; Wei, M.C.; Dowdy, S.F. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc. Natl. Acad. Sci. USA 1997, 94, 10699–10704. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.; Oakes, V.; Beamish, H.; Giles, N.; Stevens, F.; Somodevilla-Torres, M.; DeSouza, C.; Gabrielli, B. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008, 27, 4261–4268. [Google Scholar] [CrossRef]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 Inhibitors Expand the Therapeutic Options in Breast Cancer: Palbociclib, Ribociclib and Abemaciclib. BioDrugs 2019, 33, 125–135. [Google Scholar] [CrossRef]
- George, M.A.; Qureshi, S.; Omene, C.; Toppmeyer, D.L.; Ganesan, S. Clinical and Pharmacologic Differences of CDK4/6 Inhibitors in Breast Cancer. Front. Oncol. 2021, 11, 693104. [Google Scholar] [CrossRef]
- Liu, M.; Liu, H.; Chen, J. Mechanisms of the CDK4/6 inhibitor palbociclib (PD 0332991) and its future application in cancer treatment (Review). Oncol. Rep. 2018, 39, 901–911. [Google Scholar] [CrossRef]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance. Int. J. Cancer 2018, 142, 2139–2152. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, Q.; Hu, R.; Li, Y.; Peng, Y.; Liu, H.; Ye, M.; Zhang, B.; Zhang, P.; Liu-Smith, F.; et al. Novel therapeutic strategy for melanoma based on albendazole and the CDK4/6 inhibitor palbociclib. Sci. Rep. 2022, 12, 5706. [Google Scholar] [CrossRef]
- Tanaka, R.; Squires, M.S.; Kimura, S.; Yokota, A.; Nagao, R.; Yamauchi, T.; Takeuchi, M.; Yao, H.; Reule, M.; Smyth, T.; et al. Activity of the multitargeted kinase inhibitor, AT9283, in imatinib-resistant BCR-ABL-positive leukemic cells. Blood 2010, 116, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Tsubaki, M.; Genno, S.; Nemoto, C.; Onishi, Y.; Yamamoto, Y.; Imano, M.; Satou, T.; Nishida, S. AT9283 exhibits antiproliferative effect on tyrosine kinase inhibitor-sensitive and -resistant chronic myeloid leukemia cells by inhibition of Aurora A and Aurora B. Oncol. Rep. 2020, 44, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Hideshima, T.; Cirstea, D.; Bandi, M.; Nelson, E.A.; Gorgun, G.; Rodig, S.; Vallet, S.; Pozzi, S.; Patel, K.; et al. Antimyeloma activity of a multitargeted kinase inhibitor, AT9283, via potent Aurora kinase and STAT3 inhibition either alone or in combination with lenalidomide. Clin. Cancer Res. 2011, 17, 3259–3271. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Curry, J.E.; Barber, K.; Beer, P.A.; Graham, B.; Lyons, J.F.; Richardson, C.J.; Scott, M.A.; Smyth, T.; Squires, M.S.; et al. AT9283, a potent inhibitor of the Aurora kinases and Jak2, has therapeutic potential in myeloproliferative disorders. Br. J. Haematol. 2010, 150, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Crowley, B.J.; Hudon, H.E.; Gramlich, J.L.; Neuberg, D.S.; Podar, K.; Griffin, J.D.; Sattler, M. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J. Biol. Chem. 2006, 281, 18177–18183. [Google Scholar] [CrossRef] [PubMed]
- Pophali, P.A.; Patnaik, M.M. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. 2016, 22, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Von Mehren, M. Nilotinib: A novel, selective tyrosine kinase inhibitor. Semin. Oncol. 2011, 38 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Golemovic, M.; Kantarjian, H.; Manshouri, T.; Estrov, Z.; Manley, P.; Sun, T.; Arlinghaus, R.B.; Alland, L.; Dugan, M.; et al. AMN107, a novel aminopyrimidine inhibitor of p190 Bcr-Abl activation and of in vitro proliferation of Philadelphia-positive acute lymphoblastic leukemia cells. Cancer 2005, 104, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef]
- Chow, K.U.; Nowak, D.; Trepohl, B.; Hochmuth, S.; Schneider, B.; Hoelzer, D.; Mitrou, P.S.; Bergmann, L.; Ottmann, O.G.; Boehrer, S. The tyrosine kinase inhibitor AMN107 (Nilotinib) exhibits off-target effects in lymphoblastic cell lines. Leuk. Lymphoma 2007, 48, 1379–1388. [Google Scholar] [CrossRef]
- Zippilli, C.; Filippi, S.; Cesarini, S.; Bizzarri, B.M.; Conigliaro, P.; De Marchi, E.; Botta, L.; Saladino, R. Synthesis of Artesunic Acid-Coumarin Hybrids as Potential Antimelanoma Agents. ACS Med. Chem. Lett. 2023, 14, 599–605. [Google Scholar] [CrossRef]
- Ahmed, M.; Kim, D.R. pcr: An R package for quality assessment, analysis and testing of qPCR data. PeerJ 2018, 6, e4473. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | NPA Coeff (5%, 95% CI) | Node | Perturbation (5%, 95% CI) |
|---|---|---|---|
| AT-9283 | 0.16 (0.14, 0.18) | CCNA1 | −0.19 (−0.24, −0.15) |
| CCNA2 | −0.18 (−0.24, −0.12) | ||
| CDKN1A | 0.35 (0.33, 0.36) | ||
| Rb1 | 0.36 (0.34, 0.37) | ||
| Rbl2 | 0.29 (0.27, 0.32) | ||
| Imatinib | 0.01 (0, 0.02) | CCNA1 | −0.13 (−0.2, −0.06) |
| CCNA2 | 0.12 (0.02,0.22) | ||
| CDKN1A | 0.09 (0.07, 0.12) | ||
| Rb1 | 0.08 (0.06, 0.1) | ||
| Rbl2 | 0.11 (0.07, 0.15) | ||
| Nilotinib | 0.08 (0.07, 0.09) | CCNA1 | −0.14 (−0.18. −0.1) |
| CCNA2 | −0.14 (−0.2, −0.09) | ||
| CDKN1A | 0.24 (0.23, 0.26) | ||
| Rb1 | 0.25 (0.24, 0.27) | ||
| Rbl2 | 0.22 (0.2, 0.24) | ||
| Tozasertib | 0.02 (0.02, 0.02) | CCNA1 | −0.01 (−0.03, 0.01) |
| CCNA2 | −0.02 (−0.06, 0.01) | ||
| CDKN1A | 0.12 (0.11, 0.12) | ||
| Rb1 | 0.14 (0.14, 0.15) | ||
| Rbl2 | 0.11 (0.09, 0.12) | ||
| ZM-306416 | 0.02 (0.01, 0.03) | CCNA1 | −0.08 (−0.16, -0.01) |
| CCNA2 | −0.12 (−0.23, −0.01) | ||
| CDKN1A | 0.1 (0.07, 0.13) | ||
| Rb1 | 0.08 (0.06, 0.11) | ||
| Rbl2 | 0.08 (0.03, 0.12) |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | -TGCACCACCAACTGCTTAGC- | -GGCATGGACTGTGGTCATGAG- |
| CCNE1 | -GCTCCAGGAAGAGGAAGGCA- | -ATTGTCCCAAGGCTGGCTCC- |
| CCNA2 | -AGACCTACCTCAAAGCACCACAG- | -GGTTGAGGAGAGAAACACCAT- |
| CDK2 | -GCATCTTTGCTGAGATGGTG- | TAGAAGTAACTCCTGGCCAC- |
| POLA1 | -CAGTTTTGGGCTGGTTGGCG- | -GCACTCGCCCCTATCTCACA- |
| TK-1 | -GGGGCAGATCCAGGTGATTC- | -CCATGGTGTTCCGGTCATGT-3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, T.M.; Ahmed, M.; Lai, T.H.; Bahar, M.E.; Hwang, J.S.; Maulidi, R.F.; Ngo, Q.N.; Kim, D.R. Regulation of Cell Cycle Progression through RB Phosphorylation by Nilotinib and AT-9283 in Human Melanoma A375P Cells. Int. J. Mol. Sci. 2024, 25, 2956. https://doi.org/10.3390/ijms25052956
Pham TM, Ahmed M, Lai TH, Bahar ME, Hwang JS, Maulidi RF, Ngo QN, Kim DR. Regulation of Cell Cycle Progression through RB Phosphorylation by Nilotinib and AT-9283 in Human Melanoma A375P Cells. International Journal of Molecular Sciences. 2024; 25(5):2956. https://doi.org/10.3390/ijms25052956
Chicago/Turabian StylePham, Trang Minh, Mahmoud Ahmed, Trang Huyen Lai, Md Entaz Bahar, Jin Seok Hwang, Rizi Firman Maulidi, Quang Nhat Ngo, and Deok Ryong Kim. 2024. "Regulation of Cell Cycle Progression through RB Phosphorylation by Nilotinib and AT-9283 in Human Melanoma A375P Cells" International Journal of Molecular Sciences 25, no. 5: 2956. https://doi.org/10.3390/ijms25052956
APA StylePham, T. M., Ahmed, M., Lai, T. H., Bahar, M. E., Hwang, J. S., Maulidi, R. F., Ngo, Q. N., & Kim, D. R. (2024). Regulation of Cell Cycle Progression through RB Phosphorylation by Nilotinib and AT-9283 in Human Melanoma A375P Cells. International Journal of Molecular Sciences, 25(5), 2956. https://doi.org/10.3390/ijms25052956