

Gaining Insights into Key Structural Hotspots within the Allosteric Binding Pockets of Protein Kinases


Abstract
:1. Introduction
2. Types of Inhibitors
3. The Importance and Need for Allosteric Inhibitors
FDA-Approved Allosteric (Type III) Inhibitors
4. Comparison of Type III Inhibitor Targets
4.1. Binding Pocket Analysis
4.1.1. SiteMap
4.1.2. Comparison of Protein Binding Site
Binding Mode of Trametinib and Cobimetinib Inside the Allosteric Pocket of MEK1
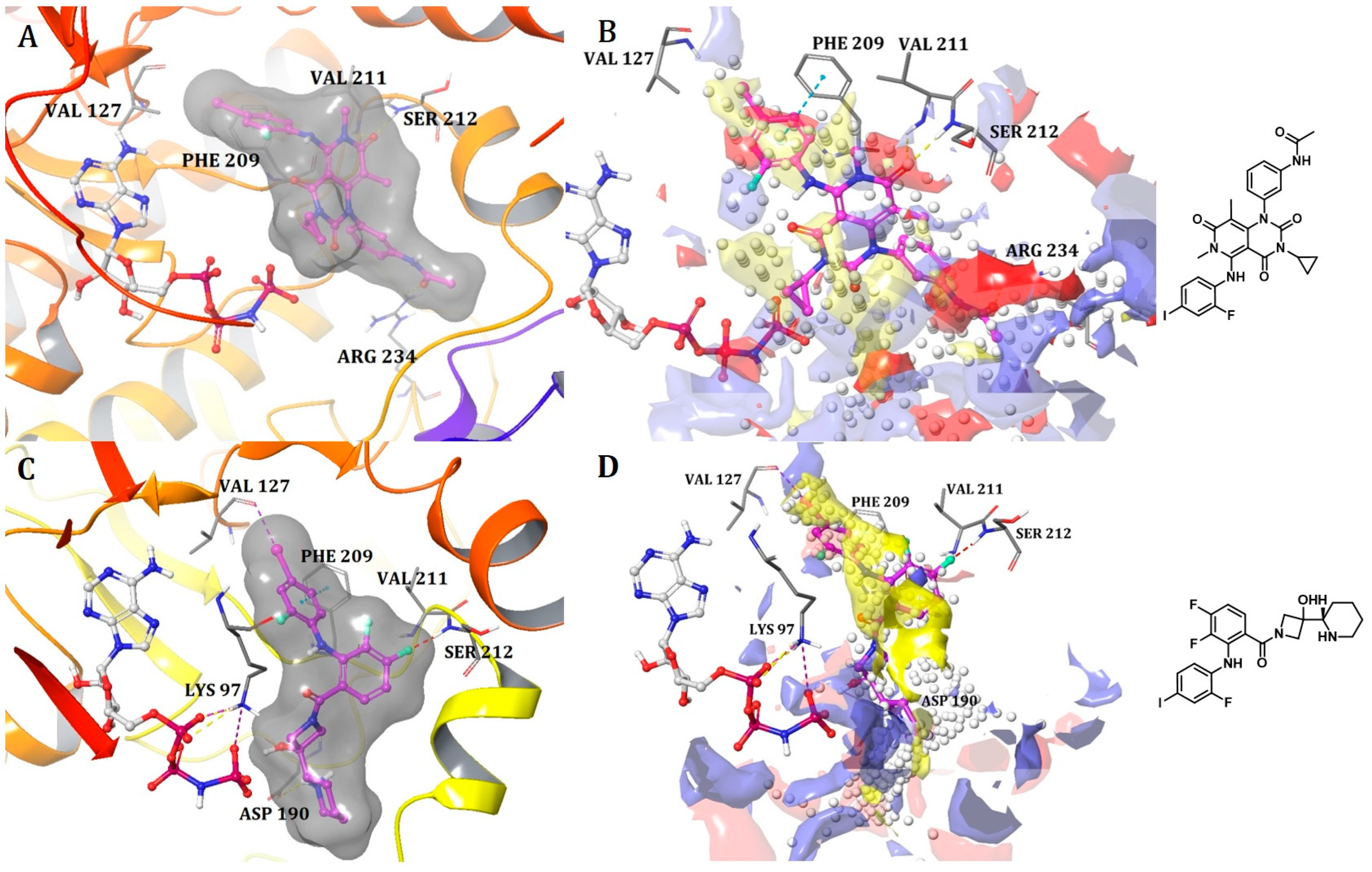
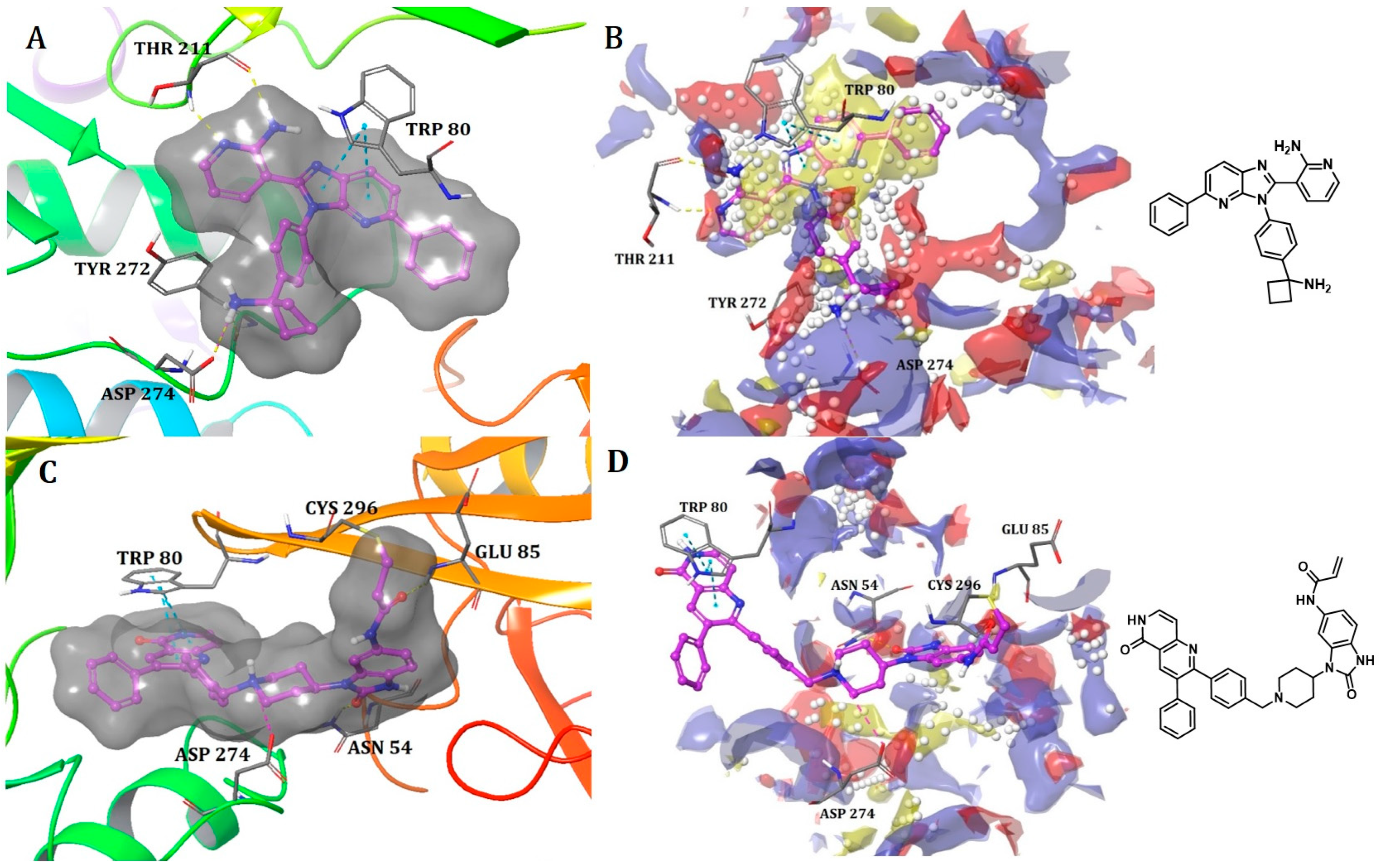
Binding Mode of ARQ092 and Borussertib Inside the Allosteric Pocket of AKT1
Binding Mode of DDC4002 and JBJ-04-125-02 Inside the Allosteric Pocket of EGFR
Binding Mode of LIMK2 with Its Co-Crystal Ligand
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sarkar, N.; Singh, A.; Kumar, P.; Kaushik, M. Protein kinases: Role of their dysregulation in carcinogenesis, identification and inhibition. Drug Res. 2023, 73, 189–199. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Wu, P.; Clausen, M.H.; Nielsen, T.E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. [Google Scholar] [CrossRef]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef]
- Salama, A.K.; Kim, K.B. MEK inhibition in the treatment of advanced melanoma. Curr. Oncol. Rep. 2013, 15, 473–482. [Google Scholar] [CrossRef]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef]
- Monod, J.; Changeux, J.-P.; Jacob, F.J. Allosteric proteins and cellular control systems. J. Mol. Biol. 1963, 6, 306–329. [Google Scholar] [CrossRef]
- Gavrin, L.K.; Saiah, E. Approaches to discover non-ATP site kinase inhibitors. MedChemComm 2013, 4, 41–51. [Google Scholar] [CrossRef]
- Fang, Z.; Grütter, C.; Rauh, D. Strategies for the selective regulation of kinases with allosteric modulators: Exploiting exclusive structural features. ACS Chem. Biol. 2013, 8, 58–70. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Pricl, S.; Kantarjian, H.; Cortes, J.; Quintás-Cardama, A. The rise and fall of gatekeeper mutations? The BCR-ABL1 T315I paradigm. Cancer 2012, 118, 293–299. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Zeiser, R. Trametinib. Small Mol. Oncol. 2014, 201, 241–248. [Google Scholar]
- Wright, C.J.; McCormack, P.L. Trametinib: First global approval. Drugs 2013, 73, 1245–1254. [Google Scholar] [CrossRef]
- Pan, Y.; Mader, M.M. Principles of kinase allosteric inhibition and pocket validation. J. Med. Chem. 2022, 65, 5288–5299. [Google Scholar] [CrossRef]
- Adjei, A.A.; LoRusso, P.; Ribas, A.; Sosman, J.A.; Pavlick, A.; Dy, G.K.; Zhou, X.; Gangolli, E.; Kneissl, M.; Faucette, S. A phase I dose-escalation study of TAK-733, an investigational oral MEK inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 47–58. [Google Scholar] [CrossRef]
- Dong, Q.; Dougan, D.R.; Gong, X.; Halkowycz, P.; Jin, B.; Kanouni, T.; O’Connell, S.M.; Scorah, N.; Shi, L.; Wallace, M.B.; et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorganic Med. Chem. Lett. 2011, 21, 1315–1319. [Google Scholar] [CrossRef]
- Martinez, R.; Defnet, A.; Shapiro, P. Avoiding or co-opting ATP inhibition: Overview of type III, IV, V, and VI kinase inhibitors. Next Gener. Kinase Inhib. 2020, 29–59. [Google Scholar] [CrossRef]
- Zhao, Z.; Xie, L.; Bourne, P.E. Insights into the binding mode of MEK type-III inhibitors. A step towards discovering and designing allosteric kinase inhibitors across the human kinome. PLoS ONE 2017, 12, e0179936. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Bhujbal, S.P.; Hah, J.-M. An Intriguing Purview on the Design of Macrocyclic Inhibitors for Unexplored Protein Kinases through Their Binding Site Comparison. Pharmaceuticals 2023, 16, 1009. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2023 update. Pharmacol. Res. 2023, 187, 106552. [Google Scholar] [CrossRef]
- Khan, Z.M.; Real, A.M.; Marsiglia, W.M.; Chow, A.; Duffy, M.E.; Yerabolu, J.R.; Scopton, A.P.; Dar, A.C. Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 2020, 588, 509–514. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.; Zak, M.; Peck, A.; Orr, C. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef]
- Lapierre, J.-M.; Eathiraj, S.; Vensel, D.; Liu, Y.; Bull, C.O.; Cornell-Kennon, S.; Iimura, S.; Kelleher, E.W.; Kizer, D.E.; Koerner, S. Discovery of 3-(3-(4-(1-Aminocyclobutyl) phenyl)-5-phenyl-3 H-imidazo [4, 5-b] pyridin-2-yl) pyridin-2-amine (ARQ 092): An orally bioavailable, selective, and potent allosteric AKT inhibitor. J. Med. Chem. 2016, 59, 6455–6469. [Google Scholar] [CrossRef]
- Weisner, J.; Landel, I.; Reintjes, C.; Uhlenbrock, N.; Trajkovic-Arsic, M.; Dienstbier, N.; Hardick, J.; Ladigan, S.; Lindemann, M.; Smith, S. Preclinical efficacy of covalent-allosteric AKT inhibitor borussertib in combination with trametinib in KRAS-mutant pancreatic and colorectal cancer. Cancer Res. 2019, 79, 2367–2378. [Google Scholar] [CrossRef]
- De Clercq, D.J.; Heppner, D.E.; To, C.; Jang, J.; Park, E.; Yun, C.-H.; Mushajiang, M.; Shin, B.H.; Gero, T.W.; Scott, D.A. Discovery and optimization of dibenzodiazepinones as allosteric mutant-selective EGFR inhibitors. ACS Med. Chem. Lett. 2019, 10, 1549–1553. [Google Scholar] [CrossRef]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef]
- Goodwin, N.C.; Cianchetta, G.; Burgoon, H.A.; Healy, J.; Mabon, R.; Strobel, E.D.; Allen, J.; Wang, S.; Hamman, B.D.; Rawlins, D.B. Discovery of a type III inhibitor of LIM kinase 2 that binds in a DFG-out conformation. ACS Med. Chem. Lett. 2015, 6, 53–57. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Protein | PDB ID | 2D Structure of Co-Crystallized Ligand | Volume of Binding Pocket (Å3) |
|---|---|---|---|---|
| 1 | MEK1 | 7JUX |  Trametinib * | 994.70 |
| 4LMN |  Cobimetinib * | 548.11 | ||
| 2 | AKT1 | 5KCV |  ARQ092 | 692.17 |
| 6HHF |  Borussertib | 418.11 | ||
| 3 | EGFR | 6P1D |  DDC4002 | 173.21 |
| 6DUK |  JBJ-04-125-02 | 416.40 | ||
| 4 | LIMK2 | 4TPT |  4TPT-ligand | 311.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhujbal, S.P.; Jun, J.; Park, H.; Moon, J.; Min, K.; Hah, J.-M. Gaining Insights into Key Structural Hotspots within the Allosteric Binding Pockets of Protein Kinases. Int. J. Mol. Sci. 2024, 25, 4725. https://doi.org/10.3390/ijms25094725
Bhujbal SP, Jun J, Park H, Moon J, Min K, Hah J-M. Gaining Insights into Key Structural Hotspots within the Allosteric Binding Pockets of Protein Kinases. International Journal of Molecular Sciences. 2024; 25(9):4725. https://doi.org/10.3390/ijms25094725
Chicago/Turabian StyleBhujbal, Swapnil P., Joonhong Jun, Haebeen Park, Jihyun Moon, Kyungbae Min, and Jung-Mi Hah. 2024. "Gaining Insights into Key Structural Hotspots within the Allosteric Binding Pockets of Protein Kinases" International Journal of Molecular Sciences 25, no. 9: 4725. https://doi.org/10.3390/ijms25094725
APA StyleBhujbal, S. P., Jun, J., Park, H., Moon, J., Min, K., & Hah, J. -M. (2024). Gaining Insights into Key Structural Hotspots within the Allosteric Binding Pockets of Protein Kinases. International Journal of Molecular Sciences, 25(9), 4725. https://doi.org/10.3390/ijms25094725