
Cytokine CCL9 Mediates Oncogenic KRAS-Induced Pancreatic Acinar-to-Ductal Metaplasia by Promoting Reactive Oxygen Species and Metalloproteinases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
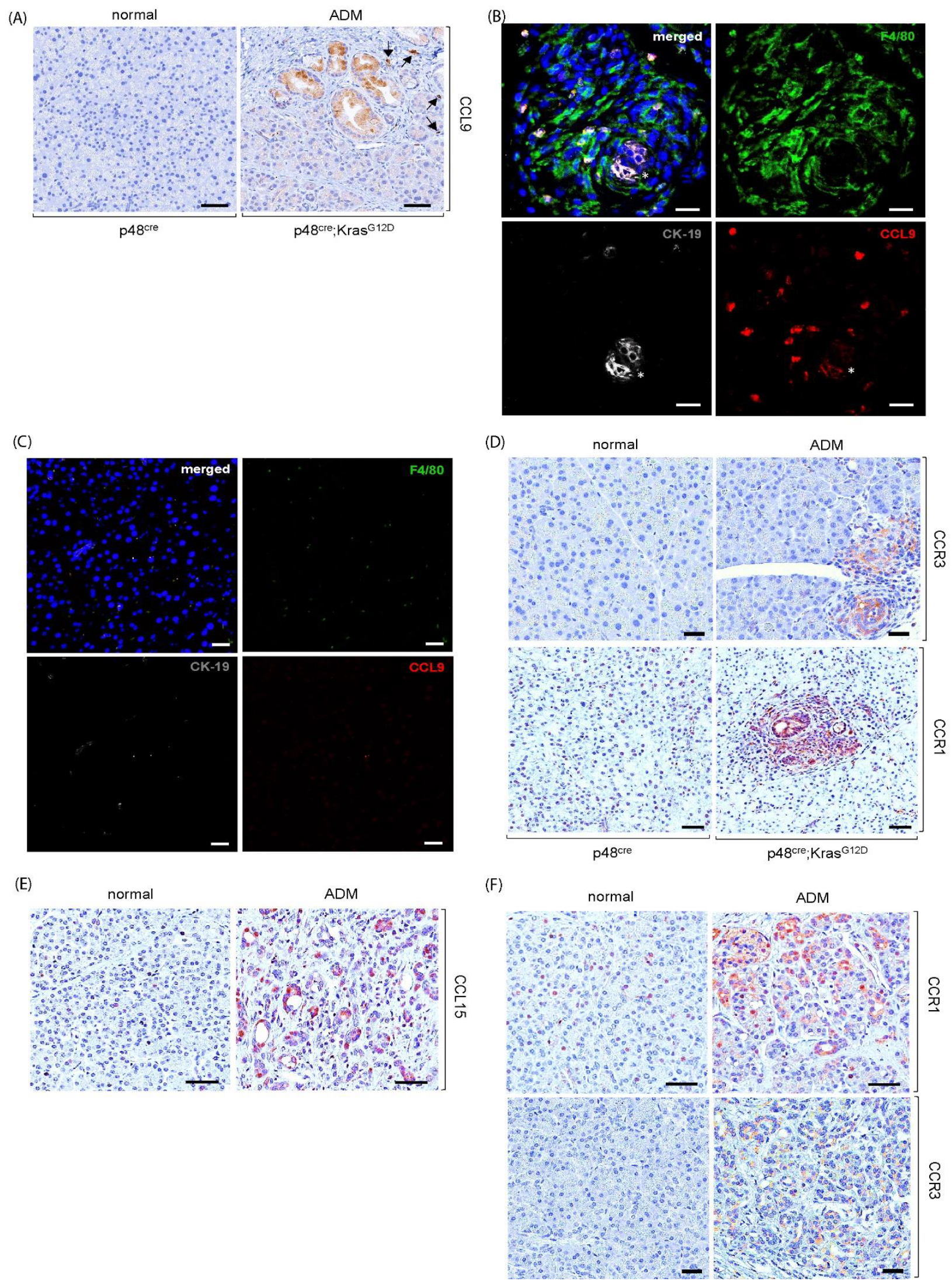
2.1. Upregulation of Cytokine CCL9 and Its Receptors in the Pancreatic ADM Regions
2.2. CCL9 Was Required for Oncogenic KrasG12D-Induced ADM of the Pancreas and a New Pancreatic ADM Driver
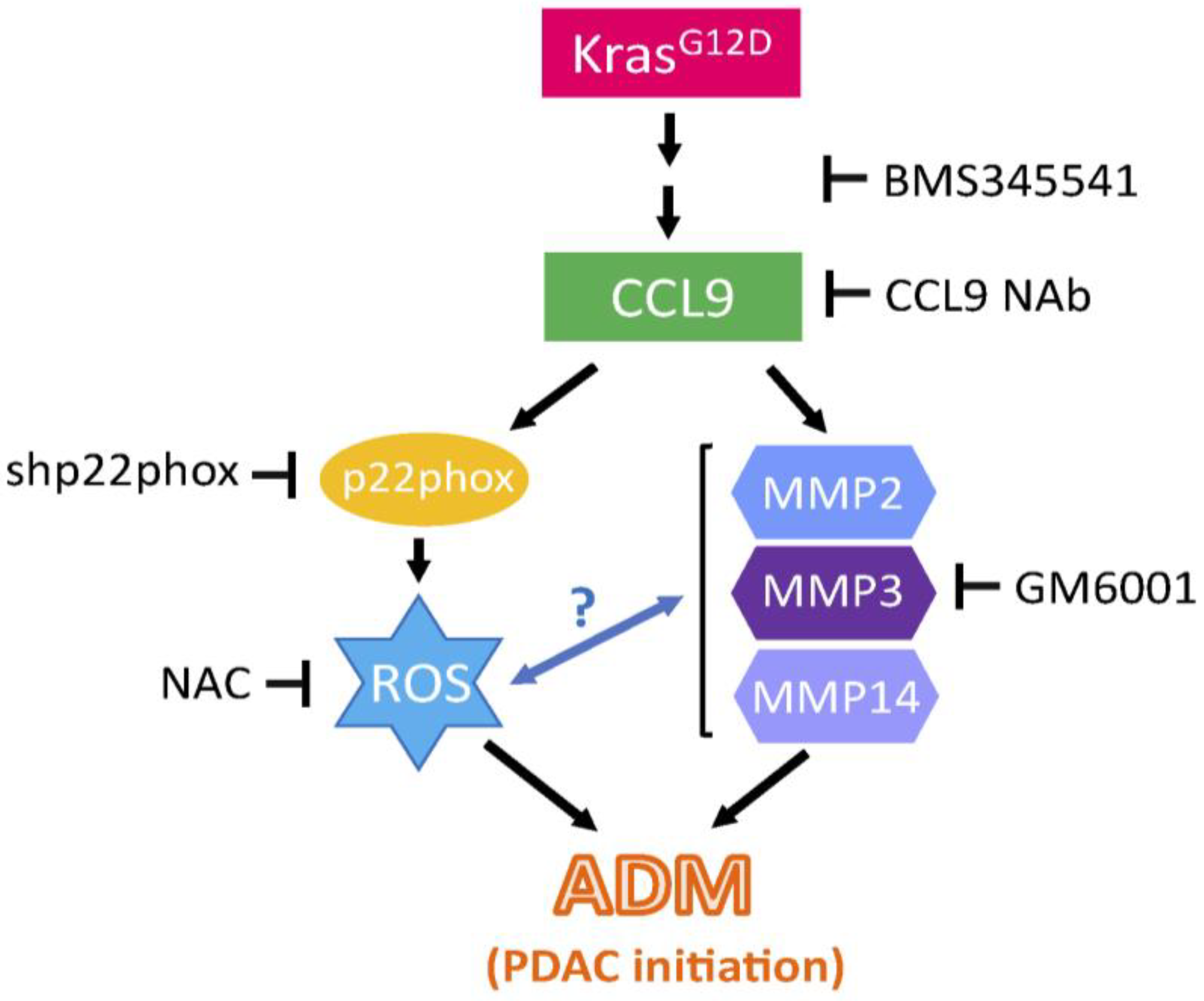
2.3. CCL9 Promoted Pancreatic ADM through Regulation of Reactive Oxygen Species (ROS)
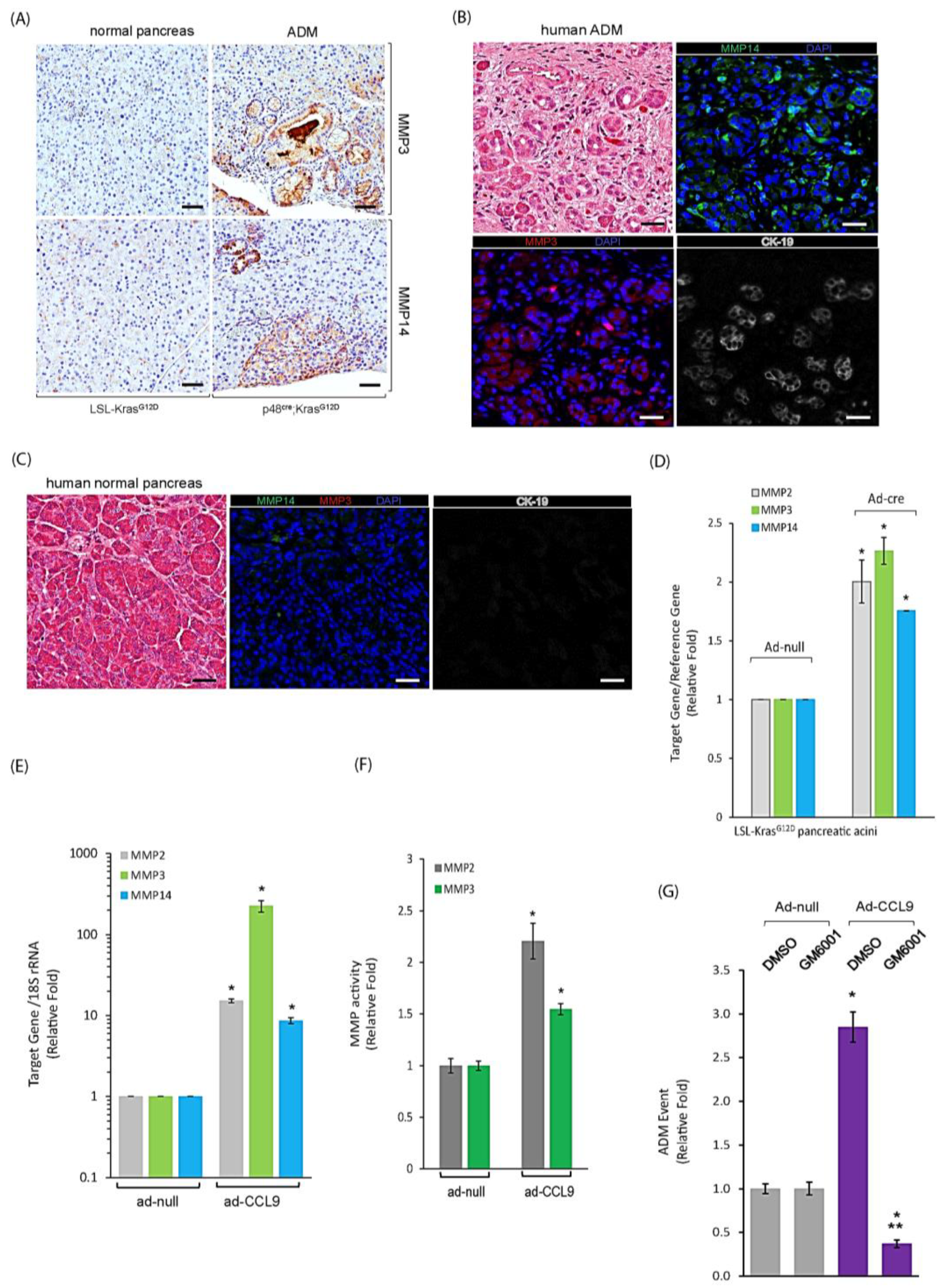
2.4. CCL9 Upregulated Metalloproteinases (MMPs) including MMP14, MMP3 and MMP2 to Modulate Pancreatic ADM
2.5. Blockade of CCL9 Diminished Oncogenic KrasG12D-Mediated Pancreatic ADM and Reduced Local Inflammation and MMPs
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Isolation and Cell Culture of Primary Pancreatic Acini
4.3. Acinar-to-Ductal Metaplasia Assay in 3D
4.4. Gene Knockdown and Overexpression Using a Viral Delivery System
4.5. RNA Isolation and Real-Time Quantitative RT-PCR
4.6. Mouse Strains and Treatment
4.7. Human Pancreas Tissue Samples
4.8. Immunohistochemistry
4.9. Measurement of MMP Activity
4.10. Measurement of Cellular ROS Levels
4.11. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Byrd, C.J. Diagnostic Bioliquid Markers for Pancreatic Cancer: What We Have vs. What We Need. Cancers 2023, 15, 2446. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol 2021, 11, 688377. [Google Scholar] [CrossRef] [PubMed]
- Sturm, N.; Ettrich, T.J.; Perkhofer, L. The Impact of Biomarkers in Pancreatic Ductal Adenocarcinoma on Diagnosis, Surveillance and Therapy. Cancers 2022, 14, 217. [Google Scholar] [CrossRef]
- Takikawa, T.; Kikuta, K.; Hamada, S.; Kume, K.; Miura, S.; Yoshida, N.; Tanaka, Y.; Matsumoto, R.; Ikeda, M.; Kataoka, F.; et al. Clinical features and prognostic impact of asymptomatic pancreatic cancer. Sci. Rep. 2022, 12, 4262. [Google Scholar] [CrossRef] [PubMed]
- Carriere, C.; Young, A.L.; Gunn, J.R.; Longnecker, D.S.; Korc, M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem. Biophys. Res. Commun. 2009, 382, 561–565. [Google Scholar] [CrossRef] [PubMed]
- De Lisle, R.C.; Logsdon, C.D. Pancreatic acinar cells in culture: Expression of acinar and ductal antigens in a growth-related manner. Eur. J. Cell Biol. 1990, 51, 64–75. [Google Scholar] [PubMed]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Liou, G.Y.; Doppler, H.; Braun, U.B.; Panayiotou, R.; Scotti Buzhardt, M.; Radisky, D.C.; Crawford, H.C.; Fields, A.P.; Murray, N.R.; Wang, Q.J.; et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 2015, 6, 6200. [Google Scholar] [CrossRef]
- Liou, G.Y.; Doppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Means, A.L.; Ray, K.C.; Singh, A.B.; Washington, M.K.; Whitehead, R.H.; Harris, R.C., Jr.; Wright, C.V.; Coffey, R.J., Jr.; Leach, S.D. Overexpression of heparin-binding EGF-like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology 2003, 124, 1020–1036. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.L.; Troutman, S.; Durham, A.; Kissil, J.L. Notch1 is not required for acinar-to-ductal metaplasia in a model of Kras-induced pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e52133. [Google Scholar] [CrossRef] [PubMed]
- De La, O.J.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; DePinho, R.A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2002, 2, 897–909. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galban, S.; Galban, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef]
- Ischenko, I.; D’Amico, S.; Rao, M.; Li, J.; Hayman, M.J.; Powers, S.; Petrenko, O.; Reich, N.C. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 2021, 12, 1482. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Bastea, L.; Fleming, A.; Doppler, H.; Edenfield, B.H.; Dawson, D.W.; Zhang, L.; Bardeesy, N.; Storz, P. The Presence of Interleukin-13 at Pancreatic ADM/PanIN Lesions Alters Macrophage Populations and Mediates Pancreatic Tumorigenesis. Cell Rep. 2017, 19, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Velez-Delgado, A.; Donahue, K.L.; Brown, K.L.; Du, W.; Irizarry-Negron, V.; Menjivar, R.E.; Lasse Opsahl, E.L.; Steele, N.G.; The, S.; Lazarus, J.; et al. Extrinsic KRAS Signaling Shapes the Pancreatic Microenvironment Through Fibroblast Reprogramming. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1673–1699. [Google Scholar] [CrossRef]
- Liou, G.Y.; Doppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Coulin, F.; Power, C.A.; Alouani, S.; Peitsch, M.C.; Schroeder, J.M.; Moshizuki, M.; Clark-Lewis, I.; Wells, T.N. Characterisation of macrophage inflammatory protein-5/human CC cytokine-2, a member of the macrophage-inflammatory-protein family of chemokines. Eur. J. Biochem. 1997, 248, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Forssmann, U.; Magert, H.J.; Adermann, K.; Escher, S.E.; Forssmann, W.G. Hemofiltrate CC chemokines with unique biochemical properties: HCC-1/CCL14a and HCC-2/CCL15. J. Leukoc. Biol. 2001, 70, 357–366. [Google Scholar] [CrossRef]
- Lean, J.M.; Murphy, C.; Fuller, K.; Chambers, T.J. CCL9/MIP-1gamma and its receptor CCR1 are the major chemokine ligand/receptor species expressed by osteoclasts. J. Cell Biochem. 2002, 87, 386–393. [Google Scholar] [CrossRef]
- Pease, J.E.; Wang, J.; Ponath, P.D.; Murphy, P.M. The N-terminal extracellular segments of the chemokine receptors CCR1 and CCR3 are determinants for MIP-1alpha and eotaxin binding, respectively, but a second domain is essential for efficient receptor activation. J. Biol. Chem. 1998, 273, 19972–19976. [Google Scholar] [CrossRef]
- Liou, G.Y.; Fleming, A.K.; Storz, P. Modulation of Protein Expression in Isolated Pancreatic Acini Using Adenoviral or Lentiviral Vectors. Available online: https://pancreapedia.org/tools/methods/modulation-of-protein-expression-in-isolated-pancreatic-acini-using-adenoviral-or (accessed on 1 June 2017).
- Pin, C.L.; Rukstalis, J.M.; Johnson, C.; Konieczny, S.F. The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J. Cell Biol. 2001, 155, 519–530. [Google Scholar] [CrossRef]
- Schreiber, F.S.; Deramaudt, T.B.; Brunner, T.B.; Boretti, M.I.; Gooch, K.J.; Stoffers, D.A.; Bernhard, E.J.; Rustgi, A.K. Successful growth and characterization of mouse pancreatic ductal cells: Functional properties of the Ki-RAS(G12V) oncogene. Gastroenterology 2004, 127, 250–260. [Google Scholar] [CrossRef]
- Vila, M.R.; Balague, C.; Real, F.X. Cytokeratins and mucins as molecular markers of cell differentiation and neoplastic transformation in the exocrine pancreas. Zentralbl. Pathol. 1994, 140, 225–235. [Google Scholar]
- Doppler, H.R.; Liou, G.Y.; Storz, P. Generation of Hydrogen Peroxide and Downstream Protein Kinase D1 Signaling Is a Common Feature of Inducers of Pancreatic Acinar-to-Ductal Metaplasia. Antioxidants 2022, 11, 137. [Google Scholar] [CrossRef]
- Liou, G.Y.; Doppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Pedre, B.; Barayeu, U.; Ezerina, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Sun, S.Y. N-acetylcysteine, reactive oxygen species and beyond. Cancer Biol. Ther. 2010, 9, 109–110. [Google Scholar] [CrossRef]
- Zhitkovich, A. N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem. Res. Toxicol. 2019, 32, 1318–1319. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Inflammatory macrophages in pancreatic acinar cell metaplasia and initiation of pancreatic cancer. Oncoscience 2015, 2, 247–251. [Google Scholar] [CrossRef]
- Deramaudt, T.; Rustgi, A.K. Mutant KRAS in the initiation of pancreatic cancer. Biochim. Biophys. Acta 2005, 1756, 97–101. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef]
- Collins, M.A.; Brisset, J.C.; Zhang, Y.; Bednar, F.; Pierre, J.; Heist, K.A.; Galban, C.J.; Galban, S.; di Magliano, M.P. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS ONE 2012, 7, e49707. [Google Scholar] [CrossRef]
- Messex, J.K.; Adams, K.L.A.; Hawkins, W.G.; DeNardo, D.; Bardeesy, N.; Billadeau, D.D.; Liou, G.Y. Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS. Cancers 2022, 14, 2153. [Google Scholar] [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Morton, J.P.; Jamieson, N.B.; Karim, S.A.; Athineos, D.; Ridgway, R.A.; Nixon, C.; McKay, C.J.; Carter, R.; Brunton, V.G.; Frame, M.C.; et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology 2010, 139, 586–597.e6. [Google Scholar] [CrossRef]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef]
- Shimizu, Y.; Dobashi, K. CC-chemokine CCL15 expression and possible implications for the pathogenesis of IgE-related severe asthma. Mediat. Inflamm. 2012, 2012, 475253. [Google Scholar] [CrossRef]
- Liu, L.Z.; Zhang, Z.; Zheng, B.H.; Shi, Y.; Duan, M.; Ma, L.J.; Wang, Z.C.; Dong, L.Q.; Dong, P.P.; Shi, J.Y.; et al. CCL15 Recruits Suppressive Monocytes to Facilitate Immune Escape and Disease Progression in Hepatocellular Carcinoma. Hepatology 2019, 69, 143–159. [Google Scholar] [CrossRef]
- Inamoto, S.; Itatani, Y.; Yamamoto, T.; Minamiguchi, S.; Hirai, H.; Iwamoto, M.; Hasegawa, S.; Taketo, M.M.; Sakai, Y.; Kawada, K. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin. Cancer Res. 2016, 22, 492–501. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Fujishita, T.; Kakizaki, F.; Hirai, H.; Matsumoto, T.; Iwamoto, M.; Inamoto, S.; Hatano, E.; Hasegawa, S.; et al. Loss of SMAD4 from colorectal cancer cells promotes CCL15 expression to recruit CCR1+ myeloid cells and facilitate liver metastasis. Gastroenterology 2013, 145, 1064–1075.e11. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kawada, K.; Itatani, Y.; Inamoto, S.; Okamura, R.; Iwamoto, M.; Miyamoto, E.; Chen-Yoshikawa, T.F.; Hirai, H.; Hasegawa, S.; et al. Loss of SMAD4 Promotes Lung Metastasis of Colorectal Cancer by Accumulation of CCR1+ Tumor-Associated Neutrophils through CCL15-CCR1 Axis. Clin. Cancer Res. 2017, 23, 833–844. [Google Scholar] [CrossRef]
- Liu, A.; Gammon, S.T.; Pisaneschi, F.; Boda, A.; Ager, C.R.; Piwnica-Worms, D.; Hong, D.S.; Curran, M.A. Hypoxia-activated prodrug and antiangiogenic therapies cooperatively treat pancreatic cancer but elicit immunosuppressive G-MDSC infiltration. JCI Insight 2024, 9, e169150. [Google Scholar] [CrossRef]
- Fang, Z.; Li, J.; Cao, F.; Li, F. Integration of scRNA-Seq and Bulk RNA-Seq Reveals Molecular Characterization of the Immune Microenvironment in Acute Pancreatitis. Biomolecules 2022, 13, 78. [Google Scholar] [CrossRef]
- Sakuma, Y.; Kodama, Y.; Eguchi, T.; Uza, N.; Tsuji, Y.; Shiokawa, M.; Maruno, T.; Kuriyama, K.; Nishikawa, Y.; Yamauchi, Y.; et al. Chemokine CXCL16 mediates acinar cell necrosis in cerulein induced acute pancreatitis in mice. Sci. Rep. 2018, 8, 8829. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells from Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Calderon, B.; Carrero, J.A.; Ferris, S.T.; Sojka, D.K.; Moore, L.; Epelman, S.; Murphy, K.M.; Yokoyama, W.M.; Randolph, G.J.; Unanue, E.R. The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med. 2015, 212, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Therapeutic Opportunities and Clinical Challenges. Cancers 2021, 13, 2860. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef] [PubMed]
- Bastea, L.I.; Liou, G.Y.; Pandey, V.; Fleming, A.K.; von Roemeling, C.A.; Doeppler, H.; Li, Z.; Qiu, Y.; Edenfield, B.; Copland, J.A.; et al. Pomalidomide Alters Pancreatic Macrophage Populations to Generate an Immune-Responsive Environment at Precancerous and Cancerous Lesions. Cancer Res. 2019, 79, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Loffek, S.; Schilling, O.; Franzke, C.W. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef]
- Crawford, H.C.; Scoggins, C.R.; Washington, M.K.; Matrisian, L.M.; Leach, S.D. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J. Clin. Investig. 2002, 109, 1437–1444. [Google Scholar] [CrossRef]
- Sawey, E.T.; Johnson, J.A.; Crawford, H.C. Matrix metalloproteinase 7 controls pancreatic acinar cell transdifferentiation by activating the Notch signaling pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 19327–19332. [Google Scholar] [CrossRef]
- Shin, Y.H.; Son, K.N.; Lee, G.W.; Kwon, B.S.; Kim, J. Transcriptional regulation of human CC chemokine CCL15 gene by NF-kappaB and AP-1 elements in PMA-stimulated U937 monocytoid cells. Biochim. Biophys. Acta 2005, 1732, 38–42. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liou, G.-Y.; Byrd, C.J.; Storz, P.; Messex, J.K. Cytokine CCL9 Mediates Oncogenic KRAS-Induced Pancreatic Acinar-to-Ductal Metaplasia by Promoting Reactive Oxygen Species and Metalloproteinases. Int. J. Mol. Sci. 2024, 25, 4726. https://doi.org/10.3390/ijms25094726
Liou G-Y, Byrd CJ, Storz P, Messex JK. Cytokine CCL9 Mediates Oncogenic KRAS-Induced Pancreatic Acinar-to-Ductal Metaplasia by Promoting Reactive Oxygen Species and Metalloproteinases. International Journal of Molecular Sciences. 2024; 25(9):4726. https://doi.org/10.3390/ijms25094726
Chicago/Turabian StyleLiou, Geou-Yarh, Crystal J. Byrd, Peter Storz, and Justin K. Messex. 2024. "Cytokine CCL9 Mediates Oncogenic KRAS-Induced Pancreatic Acinar-to-Ductal Metaplasia by Promoting Reactive Oxygen Species and Metalloproteinases" International Journal of Molecular Sciences 25, no. 9: 4726. https://doi.org/10.3390/ijms25094726
APA StyleLiou, G. -Y., Byrd, C. J., Storz, P., & Messex, J. K. (2024). Cytokine CCL9 Mediates Oncogenic KRAS-Induced Pancreatic Acinar-to-Ductal Metaplasia by Promoting Reactive Oxygen Species and Metalloproteinases. International Journal of Molecular Sciences, 25(9), 4726. https://doi.org/10.3390/ijms25094726