
A Comparative Investigation of the Bile Microbiome in Patients with Choledocholithiasis and Cholecystolithiasis through Metagenomic Analysis


Abstract
:1. Introduction
2. Results
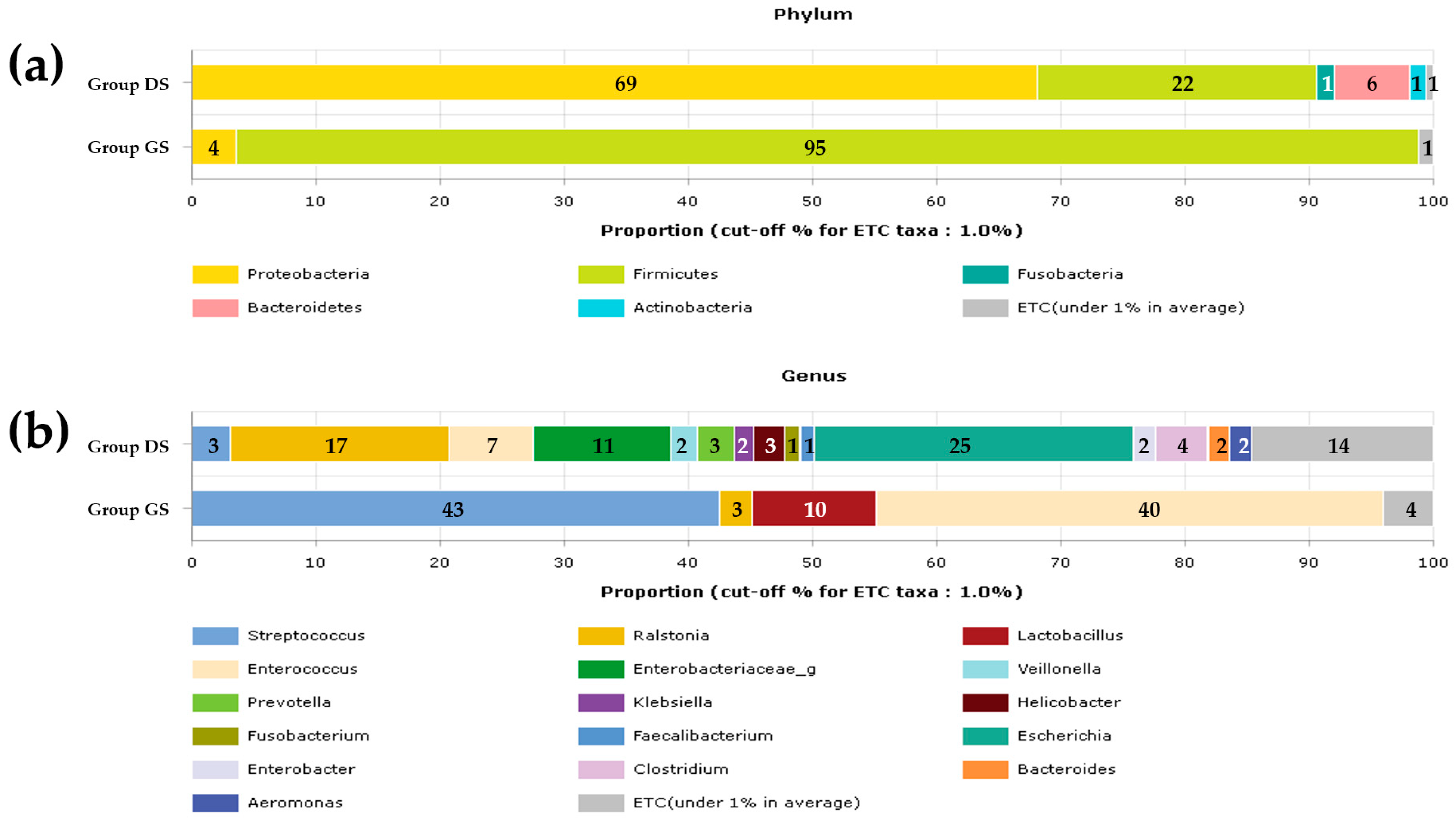
2.1. Taxonomic Composition Proportions in Bile Microbial Communities
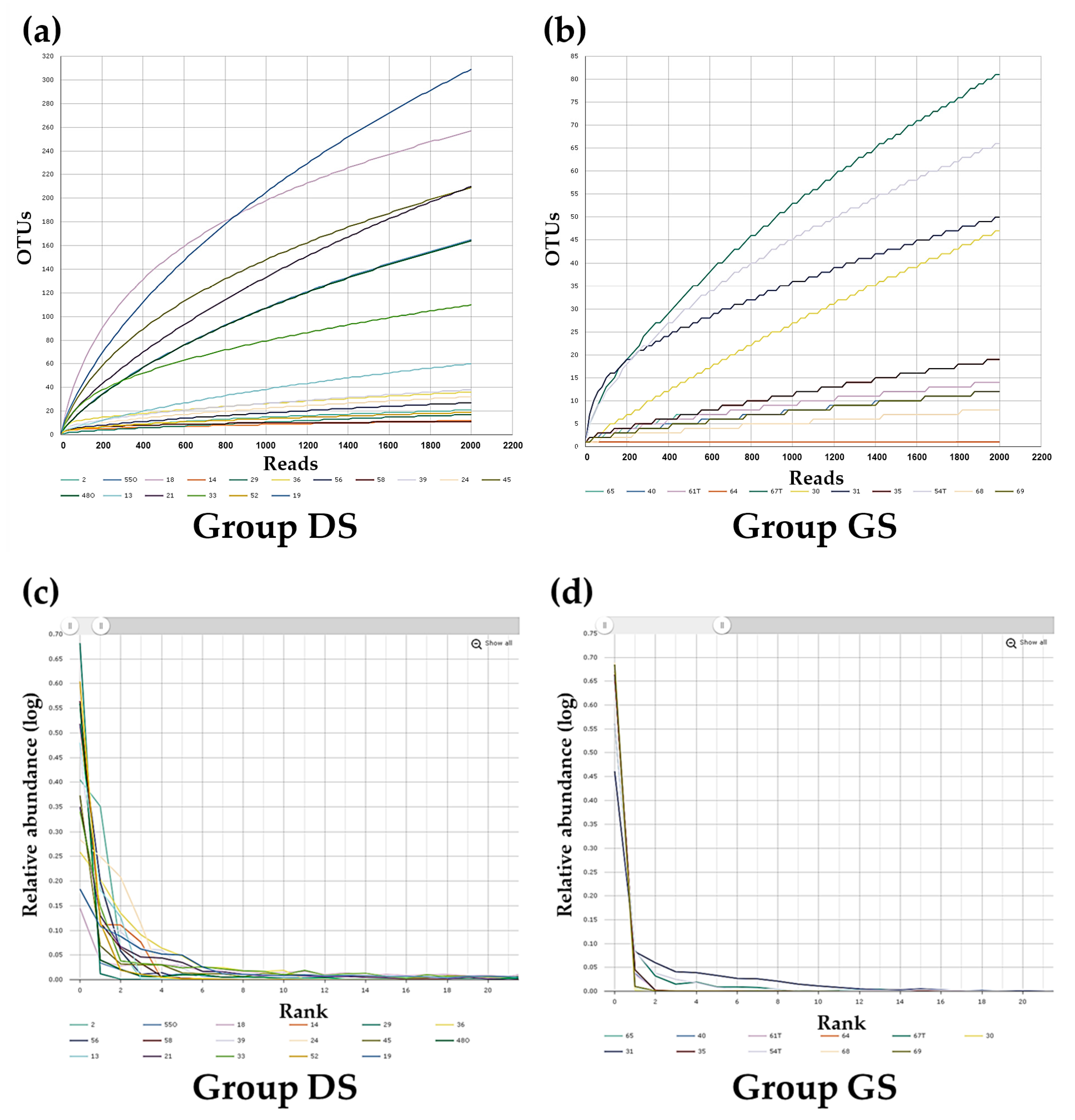
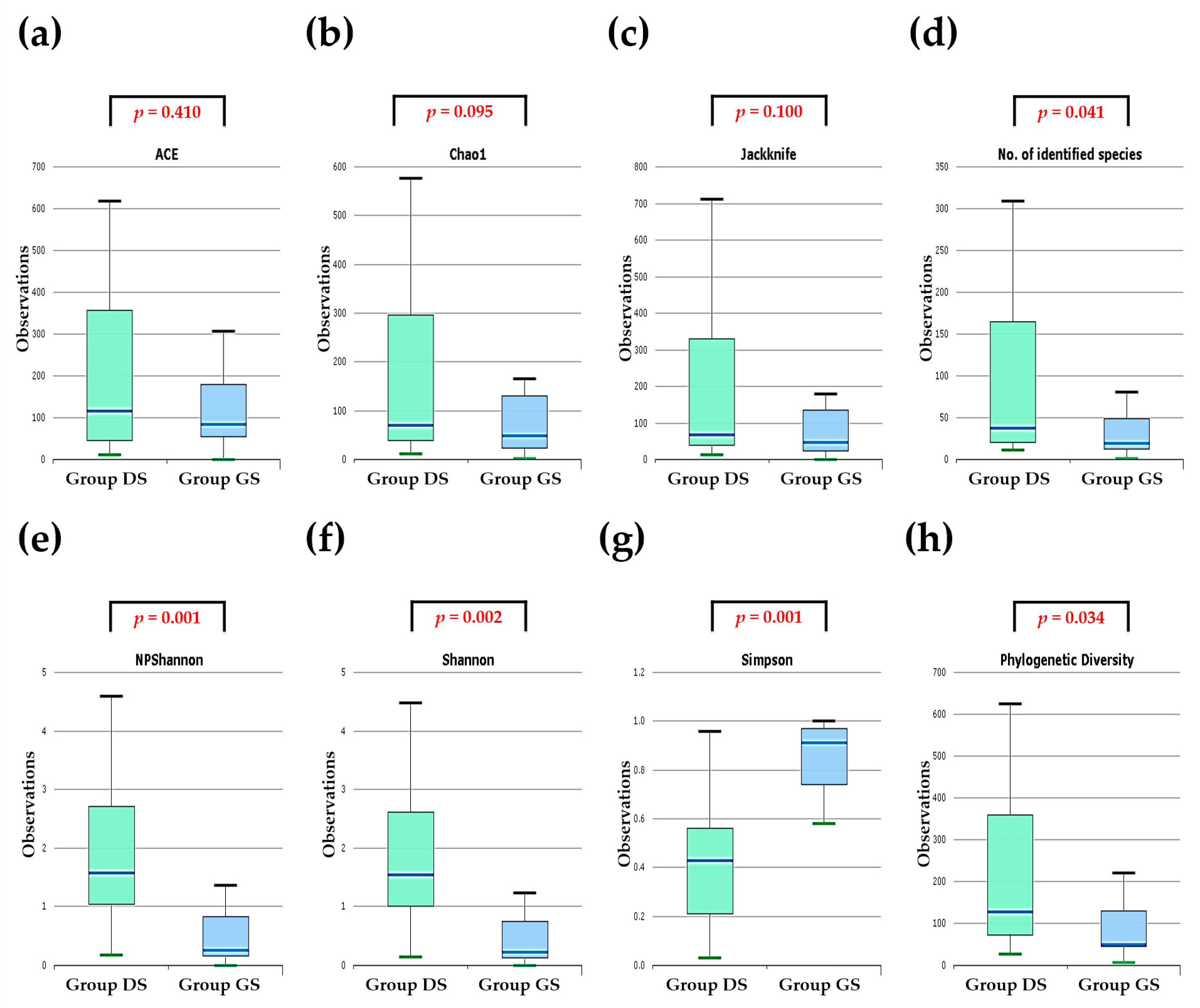
2.2. Alpha Diversity Analysis in Bile Microbial Communities
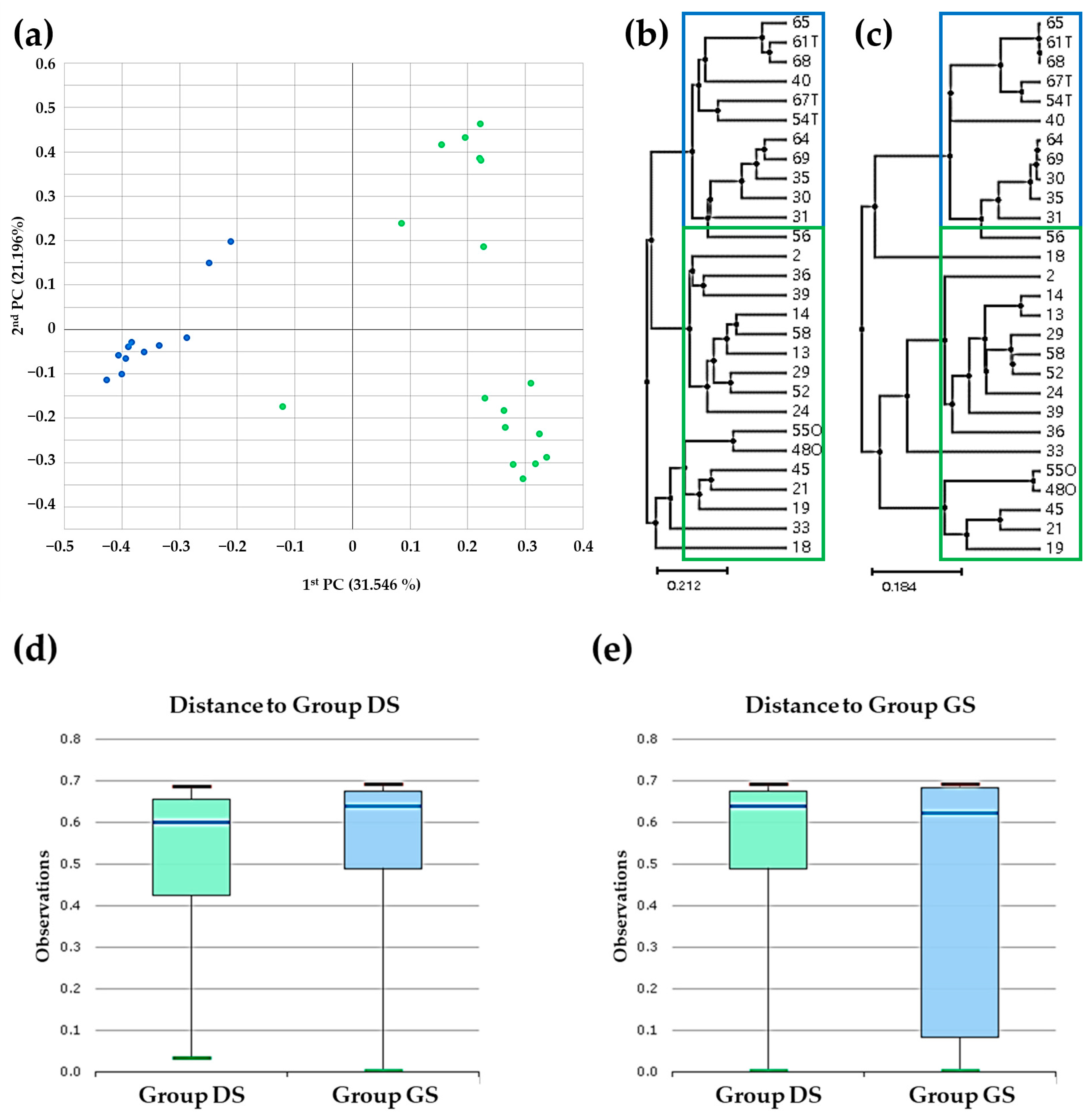
2.3. Beta Diversity Analysis in Bile Microbial Communities
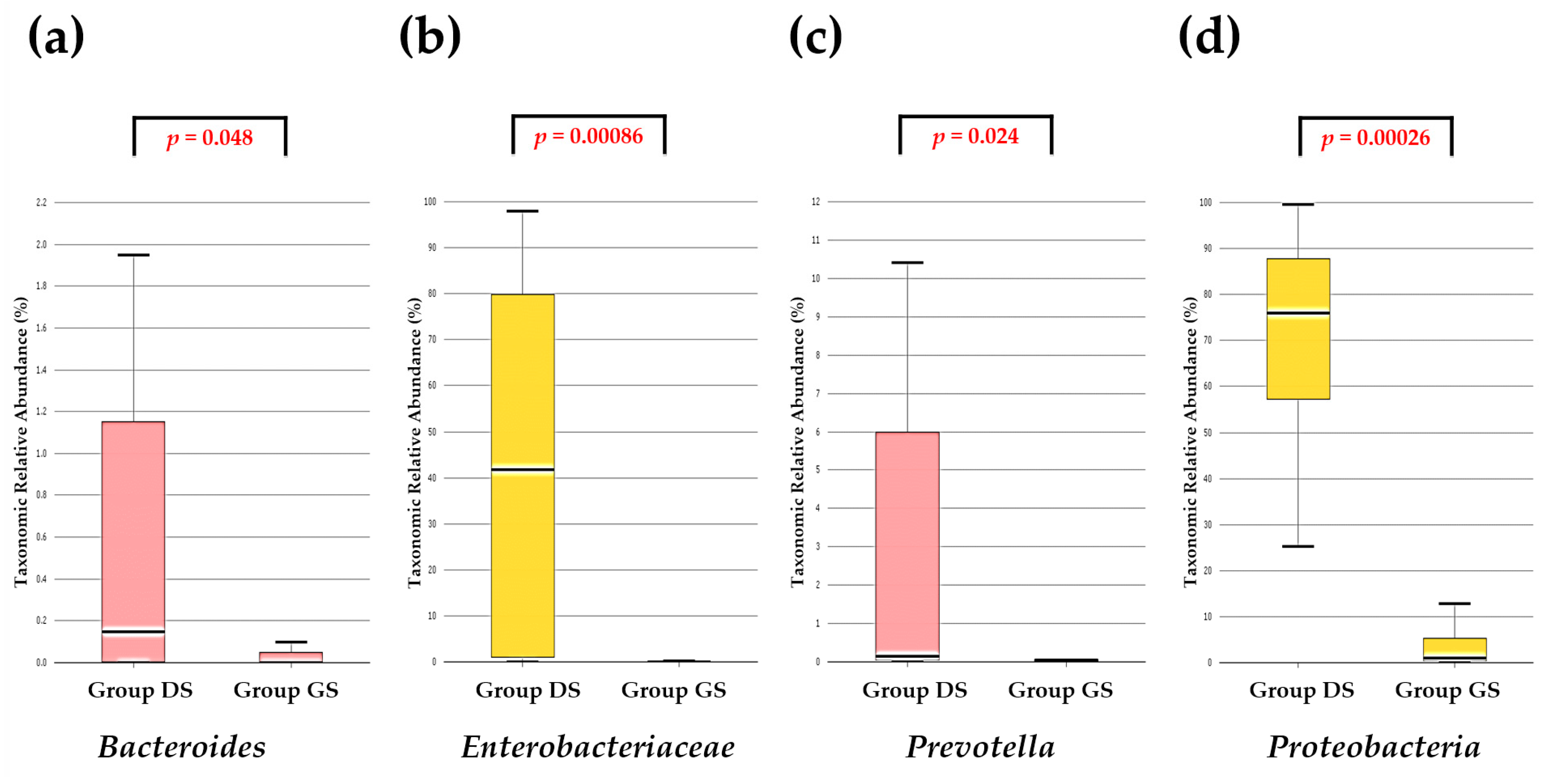
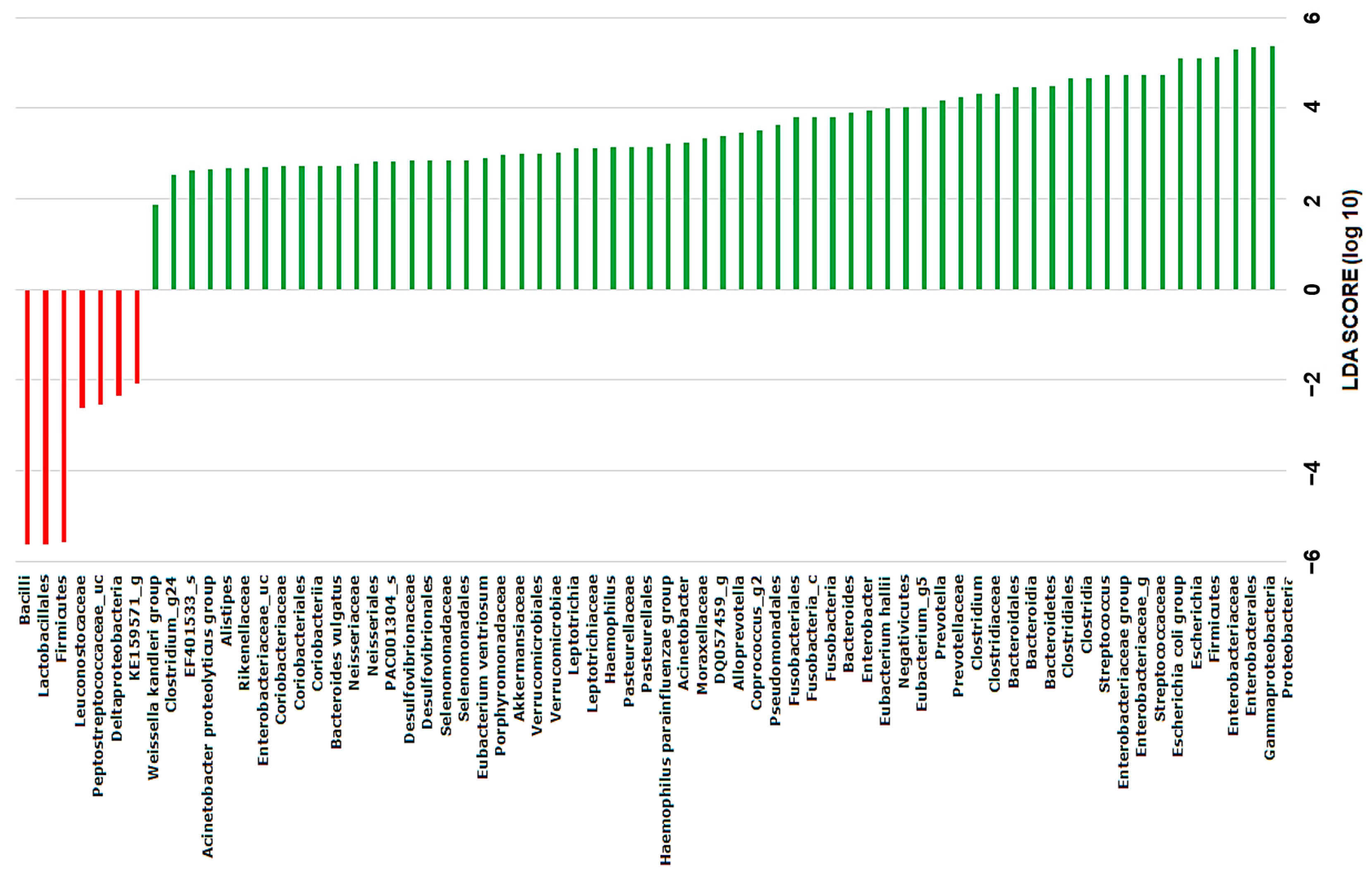
2.4. Discovery of Taxonomic Biomarkers in Bile Microbial Communities
3. Discussion
4. Materials and Methods
4.1. Specimen Collection
4.2. DNA Extraction
4.3. Massively Parallel 16S rRNA Gene Sequencing
4.4. Bioinformatic Analysis
4.5. Analysis of Taxonomic Profiling
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gurusamy, K.S.; Davidson, B.R. Gallstones. BMJ (Clin. Res. Ed.) 2014, 348, g2669. [Google Scholar] [CrossRef]
- Marschall, H.U.; Katsika, D.; Rudling, M.; Einarsson, C. The genetic background of gallstone formation: An update. Biochem. Biophys. Res. Commun. 2010, 396, 58–62. [Google Scholar] [CrossRef]
- Lammert, F.; Gurusamy, K.; Ko, C.W.; Miquel, J.F.; Mendez-Sanchez, N.; Portincasa, P.; van Erpecum, K.J.; van Laarhoven, C.J.; Wang, D.Q. Gallstones. Nature reviews. Dis. Primers 2016, 2, 16024. [Google Scholar] [CrossRef]
- Stewart, L.; Grifiss, J.M.; Jarvis, G.A.; Way, L.W. Biliary bacterial factors determine the path of gallstone formation. Am. J. Surg. 2006, 192, 598–603. [Google Scholar] [CrossRef]
- Stewart, L.; Griffiss, J.M.; Jarvis, G.A.; Way, L.W. Gallstones containing bacteria are biofilms: Bacterial slime production and ability to form pigment solids determines infection severity and bacteremia. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2007, 11, 977–983; discussion 983–974. [Google Scholar] [CrossRef]
- Shen, H.; Ye, F.; Xie, L.; Yang, J.; Li, Z.; Xu, P.; Meng, F.; Li, L.; Chen, Y.; Bo, X.; et al. Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria. Sci. Rep. 2015, 5, 17450. [Google Scholar] [CrossRef]
- Schneider, J.; De Waha, P.; Hapfelmeier, A.; Feihl, S.; Rommler, F.; Schlag, C.; Algul, H.; Schmid, R.M.; Wantia, N.; Huber, W.; et al. Risk factors for increased antimicrobial resistance: A retrospective analysis of 309 acute cholangitis episodes. J. Antimicrob. Chemother. 2014, 69, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Herzog, T.; Belyaev, O.; Akkuzu, R.; Holling, J.; Uhl, W.; Chromik, A.M. The Impact of Bile Duct Cultures on Surgical Site Infections in Pancreatic Surgery. Surg. Infect. 2015, 16, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Shen, H.; Li, Z.; Meng, F.; Li, L.; Yang, J.; Chen, Y.; Bo, X.; Zhang, X.; Ni, M. Influence of the Biliary System on Biliary Bacteria Revealed by Bacterial Communities of the Human Biliary and Upper Digestive Tracts. PLoS ONE 2016, 11, e0150519. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, J.S.; Junior, W.S.; Modena, J.L.; Brunaldi, J.E.; Ceneviva, R. Effect of preoperative endoscopic decompression on malignant biliary obstruction and postoperative infection. Hepato-Gastroenterology 2005, 52, 45–47. [Google Scholar]
- Herzog, T.; Belyaev, O.; Muller, C.A.; Mittelkotter, U.; Seelig, M.H.; Weyhe, D.; Felderbauer, P.; Schlottmann, R.; Schrader, H.; Schmidt, W.E.; et al. Bacteribilia after preoperative bile duct stenting: A prospective study. J. Clin. Gastroenterol. 2009, 43, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Stearns, J.C.; Lynch, M.D.; Senadheera, D.B.; Tenenbaum, H.C.; Goldberg, M.B.; Cvitkovitch, D.G.; Croitoru, K.; Moreno-Hagelsieb, G.; Neufeld, J.D. Bacterial biogeography of the human digestive tract. Sci. Rep. 2011, 1, 170. [Google Scholar] [CrossRef]
- Pereira, P.; Aho, V.; Arola, J.; Boyd, S.; Jokelainen, K.; Paulin, L.; Auvinen, P.; Färkkilä, M. Bile microbiota in primary sclerosing cholangitis: Impact on disease progression and development of biliary dysplasia. PLoS ONE 2017, 12, e0182924. [Google Scholar] [CrossRef]
- Feng, R.; Zhang, T.; Kayani, M.U.R.; Wang, Z.; Shen, Y.; Su, K.L.; Bielike, K.; Chen, L. Patients with Primary and Secondary Bile Duct Stones Harbor Distinct Biliary Microbial Composition and Metabolic Potential. Front. Cell. Infect. Microbiol. 2022, 12, 881489. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Chen, R.; Song, J.; He, D.; Wu, J.; Chen, X.; Yang, X.; Ye, L. Microbiota analysis with next-generation 16S rDNA gene sequencing in recurrent common bile duct stones. Ann. Transl. Med. 2022, 10, 576. [Google Scholar] [CrossRef] [PubMed]
- Molinero, N.; Ruiz, L.; Milani, C.; Gutiérrez-Díaz, I.; Sánchez, B.; Mangifesta, M.; Segura, J.; Cambero, I.; Campelo, A.B.; García-Bernardo, C.M.; et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile. Microbiome 2019, 7, 100. [Google Scholar] [CrossRef]
- Limberg, J.; Egan, C.E.; Mora, H.A.; Putzel, G.; Stamatiou, A.T.; Ullmann, T.M.; Moore, M.D.; Stefanova, D.; Thiesmeyer, J.W.; Finnerty, B.M.; et al. Metagenomic Sequencing of the Gallbladder Microbiome: Bacterial Diversity Does Not Vary by Surgical Pathology. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2022, 26, 2282–2291. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, Z.; Liu, B.; Hou, D.; Liang, Y.; Zhang, J.; Shi, P. Gut microbiota dysbiosis and bacterial community assembly associated with cholesterol gallstones in large-scale study. BMC Genom. 2013, 14, 669. [Google Scholar] [CrossRef]
- Özdirik, B.; Müller, T.; Wree, A.; Tacke, F.; Sigal, M. The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies. Int. J. Mol. Sci. 2021, 22, 6975. [Google Scholar] [CrossRef]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Grigor’eva, I.N. Gallstone Disease, Obesity and the Firmicutes/Bacteroidetes Ratio as a Possible Biomarker of Gut Dysbiosis. J. Pers. Med. 2020, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Su, W.; Zhang, Q.; Li, G.; Gao, S.; Lou, J.; Zhang, Y.; Ma, T.; Bai, X. Roles of Sphincter of Oddi Laxity in Bile Duct Microenvironment in Patients with Cholangiolithiasis: From the Perspective of the Microbiome and Metabolome. J. Am. Coll. Surg. 2016, 222, 269–280.e210. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Loza, E.; Villa-Gomez, G.; Trujillo, C.C.; Baez, S.; Asai, T.; Ikoma, T.; Endoh, K.; Nakamura, K. Metagenomics of Microbial Communities in Gallbladder Bile from Patients with Gallbladder Cancer or Cholelithiasis. Asian Pac. J. Cancer Prev. 2018, 19, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kujiraoka, M.; Kuroda, M.; Asai, K.; Sekizuka, T.; Kato, K.; Watanabe, M.; Matsukiyo, H.; Saito, T.; Ishii, T.; Katada, N.; et al. Comprehensive Diagnosis of Bacterial Infection Associated with Acute Cholecystitis Using Metagenomic Approach. Front. Microbiol. 2017, 8, 685. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hao, C.; Yao, W.; Zhu, D.; Lu, H.; Li, L.; Ma, B.; Sun, B.; Xue, D.; Zhang, W. Intestinal flora imbalance affects bile acid metabolism and is associated with gallstone formation. BMC Gastroenterol. 2020, 20, 59. [Google Scholar] [CrossRef]
- Sabater, C.; Molinero, N.; Ferrer, M.; Bernardo, C.M.G.; Delgado, S.; Margolles, A. Functional Characterisation of Bile Metagenome: Study of Metagenomic Dark Matter. Microorganisms 2021, 9, 2201. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef]
- Ye, Y.; Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group DS (n = 17) | Group GS (n = 11) | p Value | |
|---|---|---|---|
| Males/Female, n | 8/9 | 3/8 | 0.435 |
| Age, median year (range) | 67 (27–88) | 59 (30–81) | 0.456 |
| Location of gallstone, n (%) | <0.000 | ||
| Common bile duct | 10 (59%) | 0 | |
| Common bile duct and gall bladder | 7 (41%) | 0 | |
| Gall bladder | 0 | 11 (100%) | |
| Size of gallstone, n | 0.419 | ||
| Sludge | 5 (29%) | 1 (9%) | |
| <1 cm | 6 (35%) | 9 (82%) | |
| ≥1 cm | 6 (35%) | 1 (9%) | |
| Number of gallstones, n | 0.635 | ||
| Single | 7 (41%) | 8 (73%) | |
| Multiple (≥2) | 4 (24%) | 2 (18%) | |
| Not available | 6 (35%) | 1 (9%) | |
| Radiopacity of gallstone, n | 3 (18%) | 2 (18%) | 1.000 |
| Cholesterol gallstone, n | 14 (82%) | 8 (73%) | 0.653 |
| Gallstone recurrence of, n | 4 (24%) | 0 | 0.132 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, W.; Park, J. A Comparative Investigation of the Bile Microbiome in Patients with Choledocholithiasis and Cholecystolithiasis through Metagenomic Analysis. Int. J. Mol. Sci. 2024, 25, 3297. https://doi.org/10.3390/ijms25063297
Park W, Park J. A Comparative Investigation of the Bile Microbiome in Patients with Choledocholithiasis and Cholecystolithiasis through Metagenomic Analysis. International Journal of Molecular Sciences. 2024; 25(6):3297. https://doi.org/10.3390/ijms25063297
Chicago/Turabian StylePark, Wonsuk, and Joonhong Park. 2024. "A Comparative Investigation of the Bile Microbiome in Patients with Choledocholithiasis and Cholecystolithiasis through Metagenomic Analysis" International Journal of Molecular Sciences 25, no. 6: 3297. https://doi.org/10.3390/ijms25063297
APA StylePark, W., & Park, J. (2024). A Comparative Investigation of the Bile Microbiome in Patients with Choledocholithiasis and Cholecystolithiasis through Metagenomic Analysis. International Journal of Molecular Sciences, 25(6), 3297. https://doi.org/10.3390/ijms25063297