
The MDM2–p53 Axis Represents a Therapeutic Vulnerability Unique to Glioma Stem Cells
 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Differential Expression of MDM2 in Glioma Stem Cells and Non-Stem Glioma Cells
2.2. Glioma Stem Cells Are Dependent on MDM2 Expression to Prevent p53 Activation and Apoptotic Cell Death
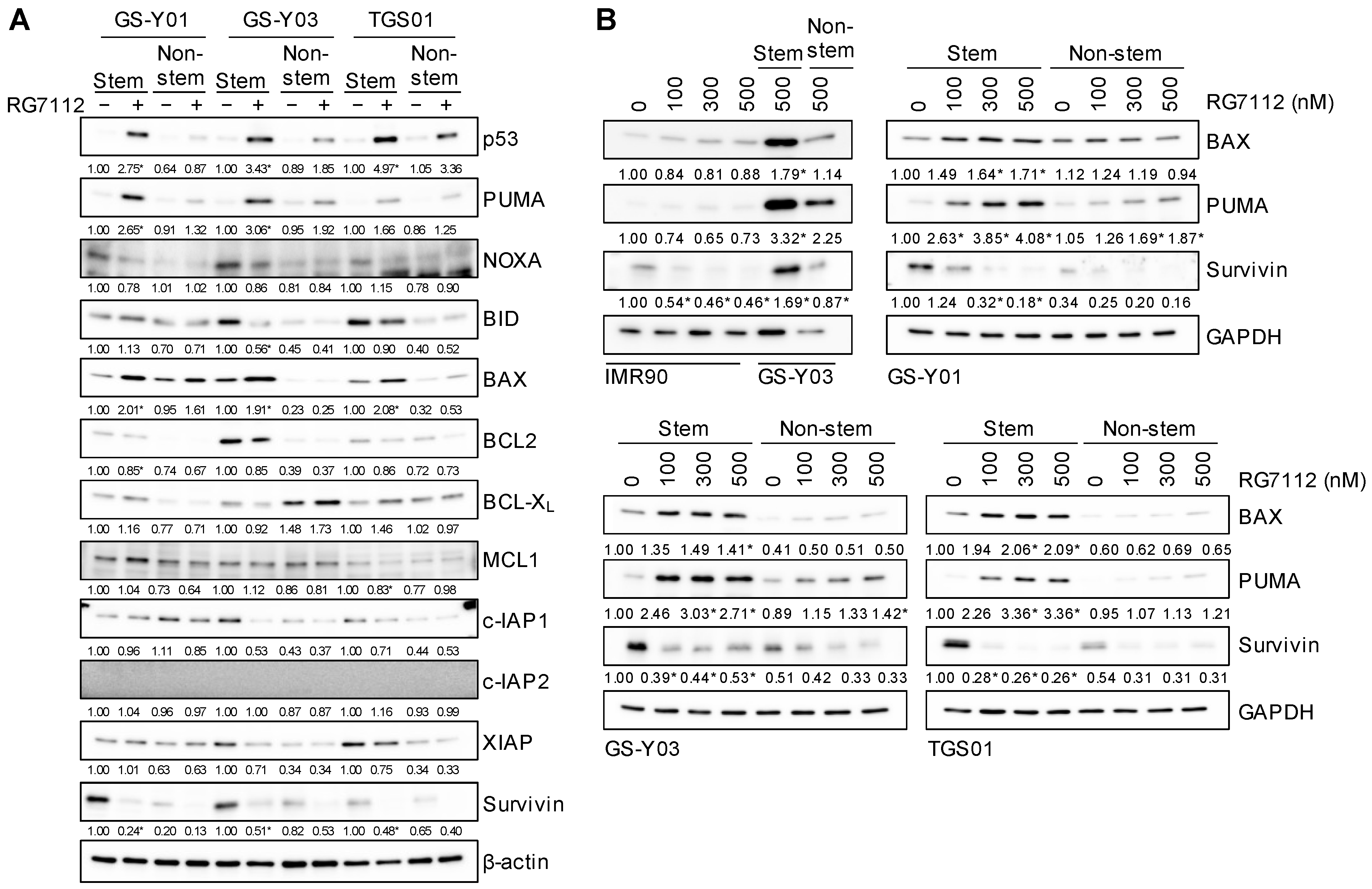
2.3. Pharmacological Inhibition of MDM2 Induces p53 Expression and Apoptotic Cell Death Preferentially in Glioma Stem Cells
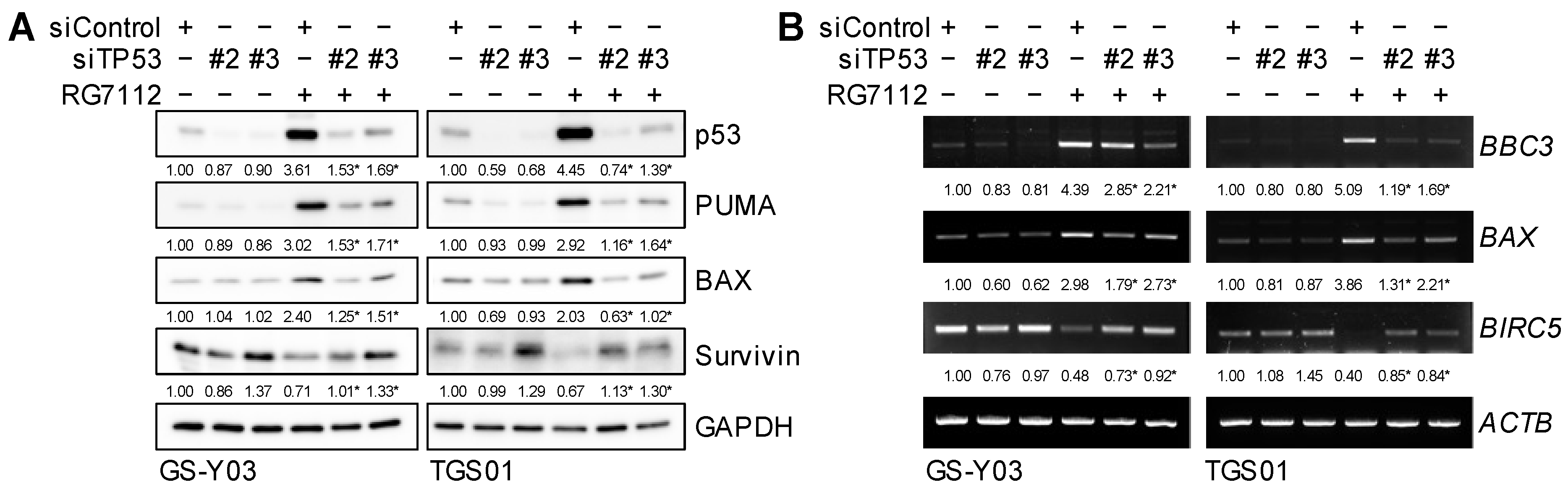
2.4. MDM2 Inhibition Induces Apoptotic Death in Glioma Stem Cells in a p53-Dependent Manner
2.5. Increased Expression of BAX and PUMA and Decreased Expression of Survivin upon MDM2 Inhibition in Glioma Stem Cells
2.6. p53-Dependent Changes in BAX, PUMA, and Survivin Expression after the Inhibition of MDM2
2.7. BAX and PUMA Expression Is Required for the Apoptotic Death of Glioma Stem Cells Induced by the Inhibition of MDM2
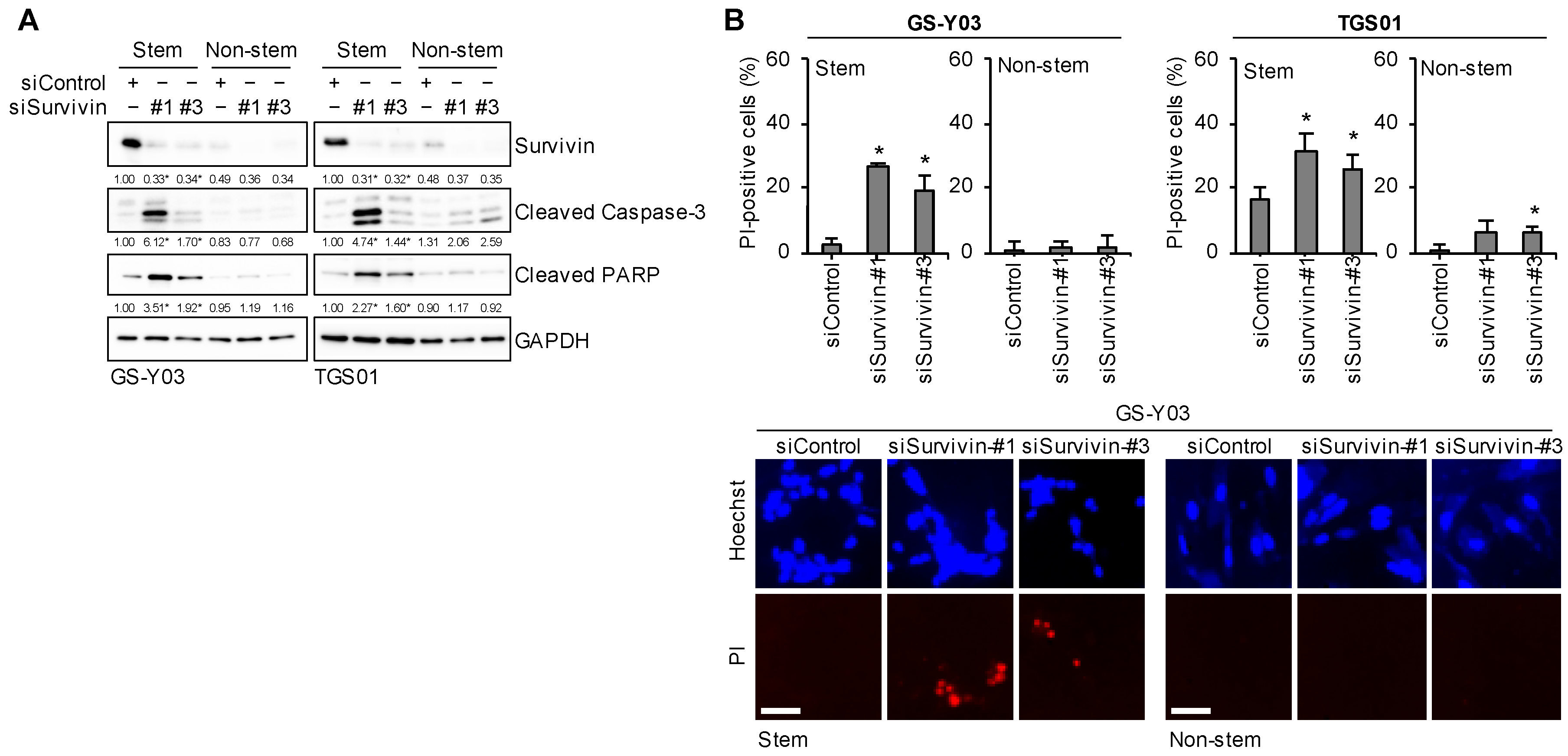
2.8. Survivin Expression Is Specifically Required for Glioma Stem Cells to Prevent Apoptotic Death
2.9. The Forced Overexpression of Survivin Protects Glioma Stem Cells from Undergoing Apoptotic Death Induced by the Inhibition of MDM2
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines, Cell Culture, and Establishment of a Glioma Stem Cell Subline Stably Overexpressing Survivin
4.3. Cell Viability/Death Assay
4.4. Western Blot Analysis
4.5. Reverse Transcription (RT)-PCR Analysis
4.6. Gene Silencing by siRNA
4.7. Data Reproducibility and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fine, H.A. New strategies in glioblastoma: Exploiting the new biology. Clin. Cancer Res. 2015, 21, 1984–1988. [Google Scholar] [CrossRef]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Karschnia, P.; Young, J.S.; Dono, A.; Häni, L.; Sciortino, T.; Bruno, F.; Juenger, S.T.; Teske, N.; Morshed, R.A.; Haddad, A.F.; et al. Prognostic validation of a new classification system for extent of resection in glioblastoma: A report of the RANO resect group. Neuro-Oncology 2023, 25, 940–954. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458. [Google Scholar] [CrossRef]
- Gillespie, M.S.; Ward, C.M.; Davies, C.C. DNA Repair and Therapeutic Strategies in Cancer Stem Cells. Cancers 2023, 15, 1897. [Google Scholar] [CrossRef]
- Zhu, K.; Xie, V.; Huang, S. Epigenetic regulation of cancer stem cell and tumorigenesis. Adv. Cancer Res. 2020, 148, 1–26. [Google Scholar] [CrossRef]
- Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. [Google Scholar] [CrossRef]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Seymour, T.; Nowak, A.; Kakulas, F. Targeting Aggressive Cancer Stem Cells in Glioblastoma. Front. Oncol. 2015, 5, 159. [Google Scholar] [CrossRef]
- Burko, P.; D’Amico, G.; Miltykh, I.; Scalia, F.; Conway de Macario, E.; Macario, A.J.L.; Giglia, G.; Cappello, F.; Caruso Bavisotto, C. Molecular Pathways Implicated in Radioresistance of Glioblastoma Multiforme: What Is the Role of Extracellular Vesicles? Int. J. Mol. Sci. 2023, 24, 4883. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Kuramoto, K.; Suzuki, S.; Sakaki, H.; Takeda, H.; Sanomachi, T.; Seino, S.; Narita, Y.; Kayama, T.; Kitanaka, C.; Okada, M. Licochalcone A specifically induces cell death in glioma stem cells via mitochondrial dysfunction. FEBS Open Bio 2017, 7, 835–844. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef]
- Okada, M.; Suzuki, S.; Togashi, K.; Sugai, A.; Yamamoto, M.; Kitanaka, C. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. Int. J. Mol. Sci. 2021, 22, 11633. [Google Scholar] [CrossRef]
- Sighel, D.; Notarangelo, M.; Aibara, S.; Re, A.; Ricci, G.; Guida, M.; Soldano, A.; Adami, V.; Ambrosini, C.; Broso, F.; et al. Inhibition of mitochondrial translation suppresses glioblastoma stem cell growth. Cell Rep. 2021, 35, 109024. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Tuval, A.; Strandgren, C.; Heldin, A.; Palomar-Siles, M.; Wiman, K.G. Pharmacological reactivation of p53 in the era of precision anticancer medicine. Nat. Rev. Clin. Oncol. 2024, 21, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussière, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef]
- Mitobe, Y.; Suzuki, S.; Nakagawa-Saito, Y.; Togashi, K.; Sugai, A.; Sonoda, Y.; Kitanaka, C.; Okada, M. Antagonizing MDM2 Overexpression Induced by MDM4 Inhibitor CEP-1347 Effectively Reactivates Wild-Type p53 in Malignant Brain Tumor Cells. Cancers 2023, 15, 4326. [Google Scholar] [CrossRef]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Zhan, Q.; Fan, S.; Bae, I.; Guillouf, C.; Liebermann, D.A.; O’Connor, P.M.; Fornace, A.J., Jr. Induction of bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene 1994, 9, 3743–3751. [Google Scholar]
- Han, J.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 2001, 98, 11318–11323. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Liu, T.; Samadashwily, G.; Li, F.; Grossman, D. Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis 2008, 29, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Gu, L.; Findley, H.W.; Chen, C.; Dong, J.T.; Yang, L.; Zhou, M. KLF5 Interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. J. Biol. Chem. 2006, 281, 14711–14718. [Google Scholar] [CrossRef] [PubMed]
- Warrier, N.M.; Krishnan, R.K.; Prabhu, V.; Hariharapura, R.C.; Agarwal, P.; Kumar, P. Survivin Inhibition by Piperine Sensitizes Glioblastoma Cancer Stem Cells and Leads to Better Drug Response. Int. J. Mol. Sci. 2022, 23, 7604. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. Brexpiprazole, a Serotonin-Dopamine Activity Modulator, Can Sensitize Glioma Stem Cells to Osimertinib, a Third-Generation EGFR-TKI, via Survivin Reduction. Cancers 2019, 11, 947. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Regulation of apoptosis pathways in cancer stem cells. Cancer Lett. 2013, 338, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Bakyt, L.; Akhmetkaliyev, A.; Toktarkhanova, D.; Bulanin, D. Re-Sensitizing Cancer Stem Cells to Conventional Chemotherapy Agents. Int. J. Mol. Sci. 2023, 24, 2122. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019, 234, 116781. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med. 2019, 11, eaau4972. [Google Scholar] [CrossRef]
- Hao, Q.; Chen, J.; Lu, H.; Zhou, X. The ARTS of p53-dependent mitochondrial apoptosis. J. Mol. Cell Biol. 2023, 14, mjac074. [Google Scholar] [CrossRef] [PubMed]
- Hikisz, P.; Kiliańska, Z.M. PUMA, a critical mediator of cell death—One decade on from its discovery. Cell. Mol. Biol. Lett. 2012, 17, 646–669. [Google Scholar] [CrossRef] [PubMed]
- Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life 2022, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, B.J.; Yang, L.; Wei, L.; Buchanan, M.E.; Rachid, M.; Parise, R.A.; Beumer, J.H.; Eiseman, J.L.; Schoen, R.E.; Zhang, L.; et al. Targeting p53-dependent stem cell loss for intestinal chemoprotection. Sci. Transl. Med. 2018, 10, eaam7610. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and Tumorigenesis: Molecular Mechanisms and Therapeutic Strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Sah, N.K.; Khan, Z.; Khan, G.J.; Bisen, P.S. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006, 244, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs223826. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Hao, X.; Bahia, R.K.; Cseh, O.; Bozek, D.A.; Blake, S.; Rinnenthal, J.; Weyer-Czernilofsky, U.; Rudolph, D.; Artee Luchman, H. BI-907828, a novel potent MDM2 inhibitor, inhibits glioblastoma brain tumor stem cells in vitro and prolongs survival in orthotopic xenograft mouse models. Neuro-Oncology 2023, 25, 913–926. [Google Scholar] [CrossRef]
- Her, N.G.; Oh, J.W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.H. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018, 9, 792. [Google Scholar] [CrossRef]
- Pellot Ortiz, K.I.; Rechberger, J.S.; Nonnenbroich, L.F.; Daniels, D.J.; Sarkaria, J.N. MDM2 Inhibition in the Treatment of Glioblastoma: From Concept to Clinical Investigation. Biomedicines 2023, 11, 1879. [Google Scholar] [CrossRef]
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward | Reverse |
|---|---|---|
| MDM2 | GGTGCTGTAACCACCTCACA | TGAGTCCGATGATTCCTGCTG |
| BBC3/PUMA | TACGAGCGGCGGAGACAAG | AGCACAACAGCCTTTCCTGA |
| BAX | GCTTCAGGGTTTCATCCAGGATCGAG | TGCACAGGGCCTTGAGCACCAGTTTG |
| BIRC5/Survivin | CCTTTCTCAAGGACCACCGCATC | CGTCATCTGGCTCCCAGCCTT |
| ACTB | CCCATGCCATCCTGCGTCTG | CGTCATACTCCTGCTTGCTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa-Saito, Y.; Mitobe, Y.; Togashi, K.; Suzuki, S.; Sugai, A.; Takenouchi, S.; Nakamura, K.; Sonoda, Y.; Kitanaka, C.; Okada, M. The MDM2–p53 Axis Represents a Therapeutic Vulnerability Unique to Glioma Stem Cells. Int. J. Mol. Sci. 2024, 25, 3948. https://doi.org/10.3390/ijms25073948
Nakagawa-Saito Y, Mitobe Y, Togashi K, Suzuki S, Sugai A, Takenouchi S, Nakamura K, Sonoda Y, Kitanaka C, Okada M. The MDM2–p53 Axis Represents a Therapeutic Vulnerability Unique to Glioma Stem Cells. International Journal of Molecular Sciences. 2024; 25(7):3948. https://doi.org/10.3390/ijms25073948
Chicago/Turabian StyleNakagawa-Saito, Yurika, Yuta Mitobe, Keita Togashi, Shuhei Suzuki, Asuka Sugai, Senri Takenouchi, Kazuki Nakamura, Yukihiko Sonoda, Chifumi Kitanaka, and Masashi Okada. 2024. "The MDM2–p53 Axis Represents a Therapeutic Vulnerability Unique to Glioma Stem Cells" International Journal of Molecular Sciences 25, no. 7: 3948. https://doi.org/10.3390/ijms25073948
APA StyleNakagawa-Saito, Y., Mitobe, Y., Togashi, K., Suzuki, S., Sugai, A., Takenouchi, S., Nakamura, K., Sonoda, Y., Kitanaka, C., & Okada, M. (2024). The MDM2–p53 Axis Represents a Therapeutic Vulnerability Unique to Glioma Stem Cells. International Journal of Molecular Sciences, 25(7), 3948. https://doi.org/10.3390/ijms25073948