
Proteomic Profiling of Endothelial Cells Exposed to Mitomycin C: Key Proteins and Pathways Underlying Genotoxic Stress-Induced Endothelial Dysfunction

 , , , ,
, , , ,
Abstract
:1. Introduction
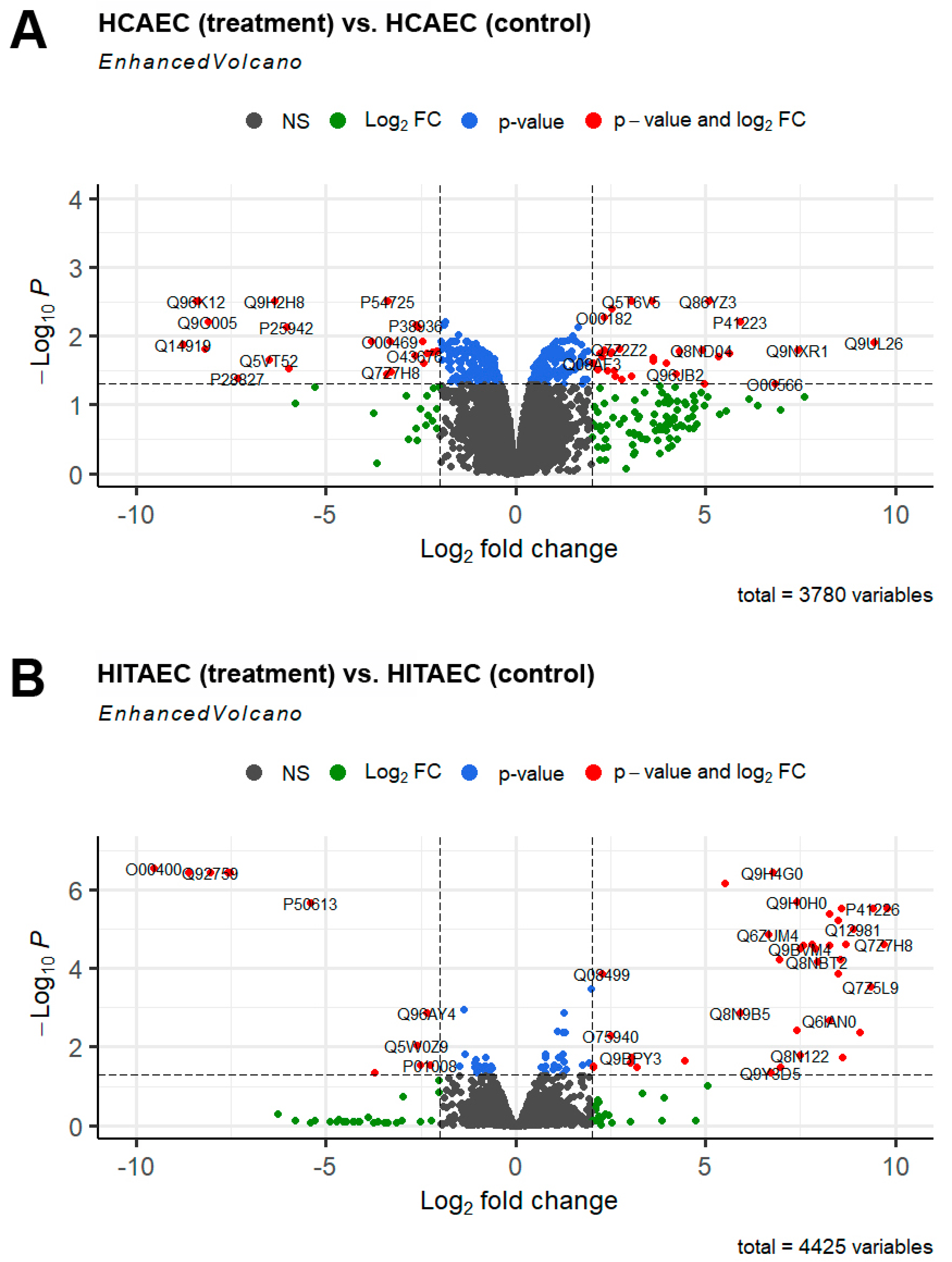
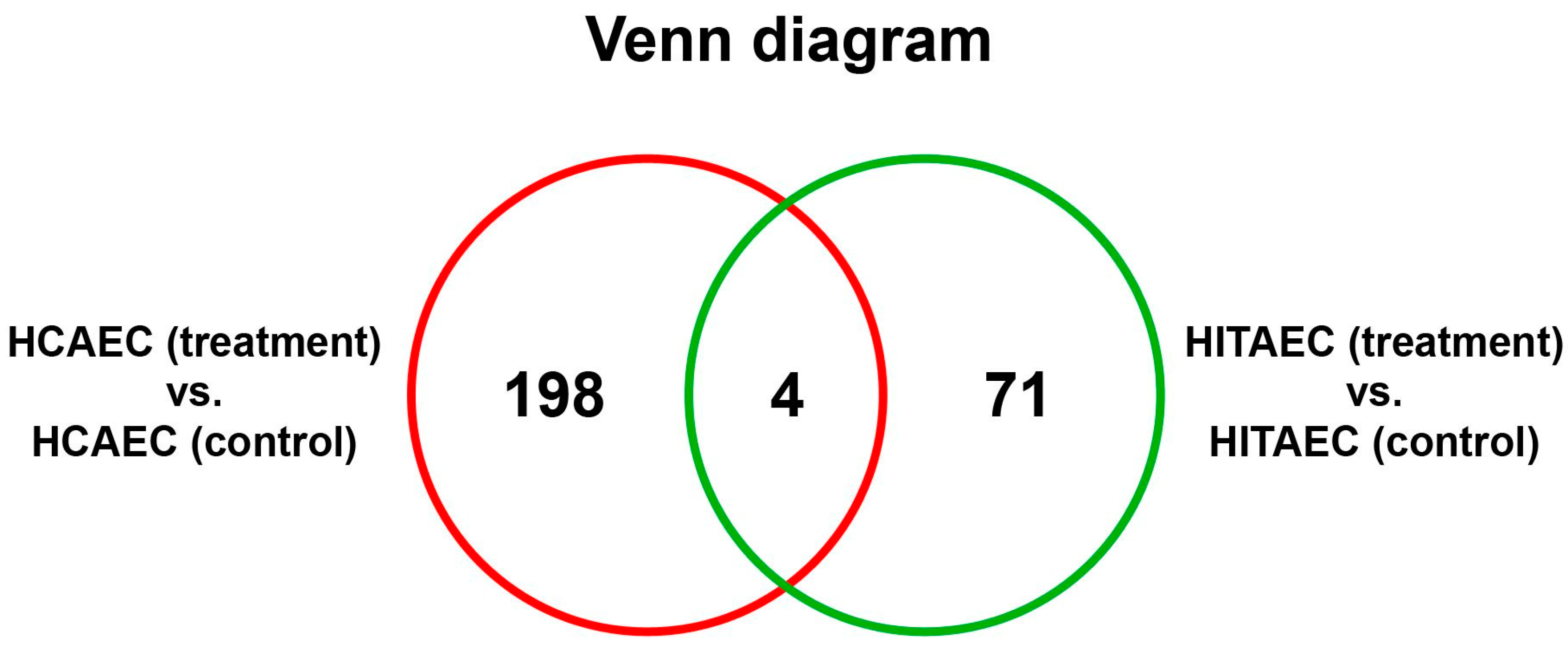
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Protein Isolation
4.3. Sample Preparation for Proteomic Profiling
4.4. Proteomic Profiling
4.5. Protein Identification
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohanan, G.; Das, A.; Rajyaguru, P.I. Genotoxic stress response: What is the role of cytoplasmic mRNA fate? BioEssays 2021, 43, e2000311. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage-how and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef]
- Hopkins, J.L.; Lan, L.; Zou, L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022, 36, 278–293. [Google Scholar] [CrossRef]
- Pulliero, A.; Godschalk, R.; Andreassi, M.G.; Curfs, D.; Van Schooten, F.J.; Izzotti, A. Environmental carcinogens and mutational pathways in atherosclerosis. Int. J. Hyg. Environ. Health 2015, 218, 293–312. [Google Scholar] [CrossRef]
- Shah, N.R.; Mahmoudi, M. The role of DNA damage and repair in atherosclerosis: A review. J. Mol. Cell. Cardiol. 2015, 86, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Niño, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.; Roks, A.J. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Sinitsky, M.Y.; Ponasenko, A.V. The role of mutagenesis in atherosclerosis. Complex Issues Cardiovasc. Dis. 2017, 1, 92–101. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Q.; Wang, T.; Long, Q.; Sun, Y.; Jiao, L.; Gullerova, M. DNA damage response, a double-edged sword for vascular aging. Ageing Res. Rev. 2023, 92, 102137. [Google Scholar] [CrossRef]
- Sinitsky, M.Y.; Kutikhin, A.G.; Tsepokina, A.V.; Shishkova, D.K.; Asanov, M.A.; Yuzhalin, A.E.; Minina, V.I.; Ponasenko, A.V. Mitomycin C induced genotoxic stress in endothelial cells is associated with differential expression of proinflammatory cytokines. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2020, 858–860, 503252. [Google Scholar] [CrossRef] [PubMed]
- Sinitsky, M.Y.; Tsepokina, A.V.; Kutikhin, A.G.; Shishkova, D.K.; Ponasenko, A.V. The gene expression profile in endothelial cells exposed to mitomycin C. Biochem. Mosc. Suppl. B Biomed. Chem. 2021, 15, 255–261. [Google Scholar] [CrossRef]
- Sinitsky, M.Y.; Sinitskaya, A.V.; Shishkova, D.K.; Ponasenko, A.V. Genotoxic stress leads to the proinflammatory response of endothelial cells: An in vitro study. Biomed. Khim. 2022, 68, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Shishkova, D.K.; Velikanova, E.A.; Sinitsky, M.Y.; Sinitskaya, A.V.; Markova, V.E. Endothelial dysfunction in the context of blood-brain barrier modeling. J. Evol. Biochem. Physiol. 2022, 58, 781–806. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.; Channon, K.M. The pathogenesis of atherosclerosis. Medicine 2014, 42, 480–484. [Google Scholar] [CrossRef]
- Bertani, F.; Di Francesco, D.; Corrado, M.D.; Talmon, M.; Fresu, L.G.; Boccafoschi, F. Paracrine Shear-Stress-Dependent Signaling from Endothelial Cells Affects Downstream Endothelial Function and Inflammation. Int. J. Mol. Sci. 2021, 22, 13300. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, J.L.; Wishnok, J.S.; Tannenbaum, S.R. Nitric oxide-induced interstrand cross-links in DNA. Chem. Res. Toxicol. 2003, 16, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Colis, L.C.; Raychaudhury, P.; Basu, A.K. Mutational specificity of gamma-radiation-induced guanine-thymine and thymineguanine intrastrand cross-links in mammalian cells and translesion synthesis past the guanine-thymine lesion by human DNA polymerase eta. Biochemistry 2008, 74, 8070–8079. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.P.; Cho, Y.J.; Huang, H.; Kim, H.Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by alpha, beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008, 41, 793–804. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A.; Medeiros, M.H.; Di Mascio, P.; Wagner, J.R. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic. Biol. Med. 2017, 107, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Sinitsky, M.; Sinitskaya, A.; Shishkova, D.; Tupikin, A.; Asanov, M.; Khutornaya, M.; Kabilov, M.; Ponasenko, A. Identification of Key Genes and Pathways in Genotoxic Stress Induced Endothelial Dysfunction: Results of Whole Transcriptome Sequencing. Biomedicines 2022, 10, 2067. [Google Scholar] [CrossRef]
- Balakrishnan, R.; Harris, M.A.; Huntley, R.; Van Auken, K.; Cherry, J.M. A guide to best practices for Gene Ontology (GO) manual annotation. Database 2013, 2013, bat054. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.A. Pathogenesis of atherosclerosis. Hematol. Am. Soc. Hematol. Educ. Program 2005, 2005, 436–441. [Google Scholar] [CrossRef]
- Sima, A.V.; Stancu, C.S.; Simionescu, M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009, 35, 191–203. [Google Scholar] [CrossRef]
- Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: AnComplex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e563–e595. [Google Scholar] [CrossRef] [PubMed]
- Branden, C.; Tooze, J. Introduction to Protein Structure, 2nd ed.; Garland Pub: New York, NY, USA, 1999; pp. 3–4. [Google Scholar]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [PubMed]
- Nakano, E.M.; Bains, H.S.; Hirai, F.E.; Portellinha, W.; Oliveira, M.; Nakano, K. Comparison of laser epithelial keratomileusis with and without mitomycin C for wavefront customized surface ablations. J. Refract. Surg. 2007, 23, S1021–S1028. [Google Scholar] [CrossRef] [PubMed]
- Pakravan, M.; Homayoon, N.; Shahin, Y.; Ali Reza, B.R. Trabeculectomy with mitomycin C versus Ahmed glaucoma implant with mitomycin C for treatment of pediatric aphakic glaucoma. J. Glaucoma 2007, 16, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Kersey, J.P.; Vivian, A.J. Mitomycin and amniotic membrane: A new method of reducing adhesions and fibrosis in strabismus surgery. Strabismus 2008, 16, 116–118. [Google Scholar] [CrossRef]
- Gupta, V.P.; Sanghi, S.; Rohatgi, J.; Dhaliwal, U. Outcomes of preoperative intrapterygial injection of mitomycin C for pterygium excision with and without inferior conjunctival flap. Oman J. Ophthalmol. 2019, 12, 171–176. [Google Scholar] [CrossRef]
- Al-Otaibi, W.A.; Alkhatib, M.H.; Wali, A.N. Cytotoxicity and apoptosis enhancement in breast and cervical cancer cells upon coadministration of mitomycin C and essential oils in nanoemulsion formulations. Biomed. Pharmacother. 2018, 106, 946–955. [Google Scholar] [CrossRef]
- Yurttas, C.; Hoffmann, G.; Tolios, A.; Haen, S.P.; Schwab, M.; Königsrainer, I.; Königsrainer, A.; Beckert, S.; Löffler, M.W. Systematic review of variations in hyperthermic intraperitoneal chemotherapy (HIPEC) for peritoneal metastasis from colorectal cancer. J. Clin. Med. 2018, 7, 567. [Google Scholar] [CrossRef] [PubMed]
- Faraj, K.; Chang, Y.H.; Rose, K.M.; Habermann, E.B.; Etzioni, D.A.; Blodgett, G.; Castle, E.P.; Humphreys, M.R.; Tyson Ii, M.D. Single-dose perioperative mitomycin-C versus thiotepa for low-grade noninvasive bladder cancer. Can. J. Urol. 2019, 26, 9922–9930. [Google Scholar] [PubMed]
- Tung, S.Y.; Lin, C.T.; Chen, C.N.; Huang, W.S. Effect of mitomycin C on X-ray repair cross complementing group 1 expression and consequent cytotoxicity regulation in human gastric cancer cells. J. Cell. Biochem. 2019, 120, 8333–8342. [Google Scholar] [CrossRef]
- Paz, M.M. Reductive activation of mitomycin C by thiols: Kinetics, mechanism, and biological implications. Chem. Res. Toxicol. 2009, 22, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Rink, S.M.; Lipman, R.; Alley, S.C.; Hopkins, P.B.; Tomasz, M. Bending of DNA by the mitomycin C-induced, GpG intrastrand cross-link. Chem. Res. Toxicol. 1996, 9, 382–389. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, S.J.; Ciccone, S.L.; Kim, C.R.; Lee, S.H. An in vivo analysis of MMC-induced DNA damage and its repair. Carcinogenesis 2006, 27, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Sakumi, K.; Fukumura, R.; Furuichi, M.; Iwasaki, Y.; Hokama, M.; Ikemura, T.; Tsuzuki, T.; Gondo, Y.; Nakabeppu, Y. 8-oxoguanine causes spontaneous de novo germline mutations in mice. Sci. Rep. 2014, 4, 4689. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, B.J.; Shield, A.J. Mutagenic damage to mammalian cells by therapeutic alkylating agents. Mutat. Res. 1996, 355, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Mildvan, A.S.; Xia, Z.; Azurmendi, H.F.; Saraswat, V.; Legler, P.M.; Massiah, M.A.; Gabelli, S.B.; Bianchet, M.A.; Kang, L.W.; Amzel, L.M. Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 2005, 433, 129–143. [Google Scholar] [CrossRef]
- McLennan, A.G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006, 63, 123–143. [Google Scholar] [CrossRef]
- Iyama, T.; Abolhassani, N.; Tsuchimoto, D.; Nonaka, M.; Nakabeppu, Y. NUDT16 is a (deoxy)inosine diphosphatase, and its deficiency induces accumulation of single-strand breaks in nuclear DNA and growth arrest. Nucleic Acids Res. 2010, 38, 4834–4843. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Ito, R.; Sekiguchi, M.; Yamagata, Y.; Setoyama, D.; Kamiya, H. Human MTH3 (NUDT18) Protein Hydrolyzes Oxidized Forms of Guanosine and Deoxyguanosine Diphosphates: Comparison with MTH1 and MTH2. J. Biol. Chem. 2012, 287, 21541–21549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kruys, V.; Huez, G.; Gueydan, C. AU-rich element-mediated translational control: Complexity and multiple activities of trans-activating factors. Biochem. Soc. Trans. 2002, 30, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, A.; Ceriani, M.C.; Capaccioli, S.; Nicolin, A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J. Cell. Physiol. 2003, 195, 356–372. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2006, 33, 7138–7150. [Google Scholar] [CrossRef] [PubMed]
- Sandler, H.; Stoecklin, G. Control of mRNA decay by phosphorylation of tristetraprolin. Biochem. Soc. Trans. 2008, 36, 491–496. [Google Scholar] [CrossRef]
- Lai, W.S.; Parker, J.S.; Grissom, S.F.; Stumpo, D.J.; Blackshear, P.J. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol. Cell. Biol. 2006, 26, 9196–9208. [Google Scholar] [CrossRef] [PubMed]
- Rappl, P.; Brüne, B.; Schmid, T. Role of Tristetraprolin in the Resolution of Inflammation. Biology 2021, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Duménil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef]
- Otsuka, S.; Ellenberg, J. Mechanisms of nuclear pore complex assembly—Two different ways of building one molecular machine. FEBS Lett. 2018, 592, 475–488. [Google Scholar] [CrossRef]
- Schöneberg, J.; Lee, I.H.; Iwasa, J.H.; Hurley, J.H. Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 2017, 18, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. ESCRT-III controls nuclear envelope reformation. Nature 2015, 522, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Schink, K.O.; Campsteijn, C.; Wegner, C.S.; Schultz, S.W.; Christ, L.; Thoresen, S.B.; Brech, A.; Raiborg, C.; Stenmark, H. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 2015, 522, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.W.; West, S.C. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat. Commun. 2014, 5, 4844. [Google Scholar] [CrossRef] [PubMed]
- Gritenaite, D.; Princz, L.N.; Szakal, B.; Bantele, S.C.; Wendeler, L.; Schilbach, S.; Habermann, B.H.; Matos, J.; Lisby, M.; Branzei, D.; et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014, 28, 1604–1619. [Google Scholar] [CrossRef] [PubMed]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Shanmugham, A.; Ovaa, H. DUBs and disease: Activity assays for inhibitor development. Curr. Opin. Drug Discov. Dev. 2008, 11, 688–696. [Google Scholar]
- Eletr, Z.M.; Wilkinson, K.D. Regulation of proteolysis by human deubiquitinating enzymes. Biochim. Biophys. Acta 2014, 1843, 114–128. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshikawa, A.; Yamagata, A.; Mimura, H.; Yamashita, M.; Ookata, K.; Nureki, O.; Iwai, K.; Komada, M.; Fukai, S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature 2008, 455, 358–362. [Google Scholar] [CrossRef]
- Ventii, K.H.; Wilkinson, K.D. Protein partners of deubiquitinating enzymes. Biochem. J. 2008, 414, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Mizushima, T.; Saeki, Y. The proteasome: Molecular machinery and pathophysiological roles. Biol. Chem. 2012, 393, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T.; Owen, D.; Harrison, S.C. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 2014, 6, a016725. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.S. Forty Years of Clathrin-coated Vesicles. Traffic 2015, 16, 1210–1238. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.K.; Sorkin, A. Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017459. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Di Fiore, P.P.; Sigismund, S. Endocytic control of signaling at the plasma membrane. Curr. Opin. Cell Biol. 2016, 39, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Traub, L.M.; Bonifacino, J.S. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016790. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Kispal, G. Mitochondrial ABC transporters. Res. Microbiol. 2001, 152, 331–340. [Google Scholar] [CrossRef]
- Liesa, M.; Qiu, W.; Shirihai, O.S. Mitochondrial ABC transporters function: The role of ABCB10 (ABC-me) as a novel player in cellular handling of reactive oxygen species. Biochim. Biophys. Acta 2012, 1823, 1945–1957. [Google Scholar] [CrossRef]
- Pahnke, J.; Fröhlich, C.; Krohn, M.; Schumacher, T.; Paarmann, K. Impaired mitochondrial energy production and ABC transporter function-A crucial interconnection in dementing proteopathies of the brain. Mech. Ageing Dev. 2013, 134, 506–515. [Google Scholar] [CrossRef]
- Jawdekar, G.W.; Henry, R.W. Transcriptional regulation of human small nuclear RNA genes. Biochim. Biophys. Acta 2008, 1779, 295–305. [Google Scholar] [CrossRef]
- Zhang, M.X.; Ou, H.; Shen, Y.H.; Wang, J.; Wang, J.; Coselli, J.; Wang, X.L. Regulation of endothelial nitric oxide synthase by small RNA. Proc. Natl. Acad. Sci. USA 2005, 102, 16967–16972. [Google Scholar] [CrossRef]
- Dvorská, D.; Braný, D.; Ňachajová, M.; Halašová, E.; Danková, Z. Breast Cancer and the Other Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 3280. [Google Scholar] [CrossRef]
- Holzer, G.; Markov, G.V.; Laudet, V. Evolution of Nuclear Receptors and Ligand Signaling: Toward a Soft Key-Lock Model? Curr. Top. Dev. Biol. 2017, 125, 1–38. [Google Scholar]
- Sims, F.H. A comparison of coronary and internal mammary arteries and implications of the results in the etiology of arteriosclerosis. Am. Heart J. 1983, 105, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Aboyans, V.; Lacroix, P.; Criqui, M.H. Large and small vessels atherosclerosis: Similarities and differences. Prog. Cardiovasc. Dis. 2007, 50, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Lobov, A.; Kabilov, M.; Zainullina, B.; Tupikin, A.; Shishkova, D.; Markova, V.; Sinitskaya, A.; Grigoriev, E.; Markova, Y.; et al. Multi-Omics Profiling of Human Endothelial Cells from the Coronary Artery and Internal Thoracic Artery Reveals Molecular but Not Functional Heterogeneity. Int. J. Mol. Sci. 2023, 24, 15032. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling, R Package Version 1.14.0; R Core Team: Vienna, Austria, 2022. [Google Scholar]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.A.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2023. R Package Version 1.20.0. Available online: https://bioconductor.org/packages/EnhancedVolcano (accessed on 1 March 2024).



{kind=link}
{kind=link}
| Pathway Name (Reactome Identifier) | Total Number of Proteins | Number of Denoted Proteins | Percent from DEPs | FDR-Corrected p-Value |
|---|---|---|---|---|
| Upregulated after MMC treatment | ||||
| Phosphate bond hydrolysis by NUDT proteins (R-HSA-2393930) | 11 | 4 | 4.3 | 6.28 × 10−4 |
| Purine catabolism (R-HSA-74259) | 28 | 4 | 4.3 | 0.012 |
| Tristetraprolin (TTP, ZFP36) binds and destabilizes mRNA (R-HSA-450513) | 258 | 8 | 8.5 | 0.041 |
| Nucleotide catabolism (R-HSA-8956319) | 46 | 4 | 4.3 | 0.041 |
| Downregulated after MMC treatment | ||||
| Sealing of the nuclear envelope (NE) by ESCRT-III (R-HSA-9668328) | 94 | 7 | 6.5 | 0.003 |
| Cargo recognition for clathrin-mediated endocytosis (R-HSA-8856825) | 157 | 8 | 7.4 | 0.004 |
| Clathrin-mediated endocytosis (R-HAS-8856828) | 213 | 9 | 8.3 | 0.004 |
| Metalloprotease DUBs (R-HSA-5689901) | 103 | 6 | 5.6 | 0.017 |
| Lysosome Vesicle Biogenesis (R-HSA-432720) | 794 | 15 | 13.9 | 0.044 |
| Trans-Golgi Network Vesicle Budding (R-HSA-199992) | 987 | 17 | 15.7 | 0.044 |
| Pathway Name | Total Number of Proteins | Number of Denoted Proteins | Percent from DEPs | FDR-Corrected p-Value |
|---|---|---|---|---|
| Downregulated after MMC treatment | ||||
| Mitochondrial ABC transporters (R-HSA-1369007) | 4 | 2 | 10.0 | 0.009 |
| RNA polymerase II transcribes snRNA genes (R-HSA-6807505) | 220 | 4 | 20.0 | 0.033 |
| NR1H2 & NR1H3 regulate gene expression linked to lipogenesis (R-HSA-9029558) | 84 | 3 | 15.5 | 0.033 |
| Molecular Term | Number of Denoted Proteins | Percent from DEPs | Fold Enrichment | FDR-Corrected p-Value |
|---|---|---|---|---|
| Upregulated after MMC treatment | ||||
| Response to platinum ion (BP) | 2 | 2.13 | >100 | 4.95 × 10−2 |
| Nucleic acid metabolic process (BP) | 24 | 25.53 | 2.41 | 3.06 × 10−2 |
| Organic substance biosynthetic process (BP) | 36 | 38.30 | 1.99 | 2.37 × 10−2 |
| Cellular component organization (BP) | 45 | 47.87 | 1.75 | 2.64 × 10−2 |
| RNA binding (MF) | 22 | 23.40 | 2.87 | 1.27 × 10−2 |
| Protein binding (MF) | 86 | 91.49 | 1.30 | 5.75 × 10−3 |
| Organelle envelope (CC) | 17 | 18.09 | 2.88 | 9.80 × 10−3 |
| Nucleoplasm (CC) | 38 | 40.43 | 2.00 | 2.33 × 10−3 |
| Cytosol (CC) | 47 | 50.00 | 1.86 | 9.30 × 10−4 |
| Protein-containing complex (CC) | 48 | 51.06 | 1.61 | 1.59 × 10−2 |
| Downregulated after MMC treatment | ||||
| Multivesicular body assembly (BP) | 5 | 4.63 | 30.64 | 5.17 × 10−3 |
| Protein metabolic process (BP) | 4 | 3.70 | 2.32 | 2.80 × 10−4 |
| Cellular biosynthetic process (BP) | 42 | 38.89 | 2.13 | 4.02 × 10−3 |
| Postsynaptic endocytic zone cytoplasmic component (CC) | 2 | 1.85 | >100 | 4.02 × 10−2 |
| ESCRT III complex (CC) | 3 | 2.78 | 53.49 | 3.22 × 10−2 |
| Amphisome membrane (CC) | 3 | 2.78 | 49.03 | 4.28 × 10−2 |
| Multivesicular body membrane (CC) | 4 | 3.70 | 27.05 | 2.15 × 10−2 |
| Clathrin vesicle coat (CC) | 4 | 3.70 | 22.41 | 4.62 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinitsky, M.; Repkin, E.; Sinitskaya, A.; Markova, V.; Shishkova, D.; Barbarash, O. Proteomic Profiling of Endothelial Cells Exposed to Mitomycin C: Key Proteins and Pathways Underlying Genotoxic Stress-Induced Endothelial Dysfunction. Int. J. Mol. Sci. 2024, 25, 4044. https://doi.org/10.3390/ijms25074044
Sinitsky M, Repkin E, Sinitskaya A, Markova V, Shishkova D, Barbarash O. Proteomic Profiling of Endothelial Cells Exposed to Mitomycin C: Key Proteins and Pathways Underlying Genotoxic Stress-Induced Endothelial Dysfunction. International Journal of Molecular Sciences. 2024; 25(7):4044. https://doi.org/10.3390/ijms25074044
Chicago/Turabian StyleSinitsky, Maxim, Egor Repkin, Anna Sinitskaya, Victoria Markova, Daria Shishkova, and Olga Barbarash. 2024. "Proteomic Profiling of Endothelial Cells Exposed to Mitomycin C: Key Proteins and Pathways Underlying Genotoxic Stress-Induced Endothelial Dysfunction" International Journal of Molecular Sciences 25, no. 7: 4044. https://doi.org/10.3390/ijms25074044
APA StyleSinitsky, M., Repkin, E., Sinitskaya, A., Markova, V., Shishkova, D., & Barbarash, O. (2024). Proteomic Profiling of Endothelial Cells Exposed to Mitomycin C: Key Proteins and Pathways Underlying Genotoxic Stress-Induced Endothelial Dysfunction. International Journal of Molecular Sciences, 25(7), 4044. https://doi.org/10.3390/ijms25074044