
The Discovery of New Inhibitors of Insulin-Regulated Aminopeptidase by a High-Throughput Screening of 400,000 Drug-like Compounds

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Development of the Screening Assay Cascade
2.1.1. Reference Compounds
2.1.2. Principle, Optimization, and Miniaturization of Primary and Orthogonal Assays
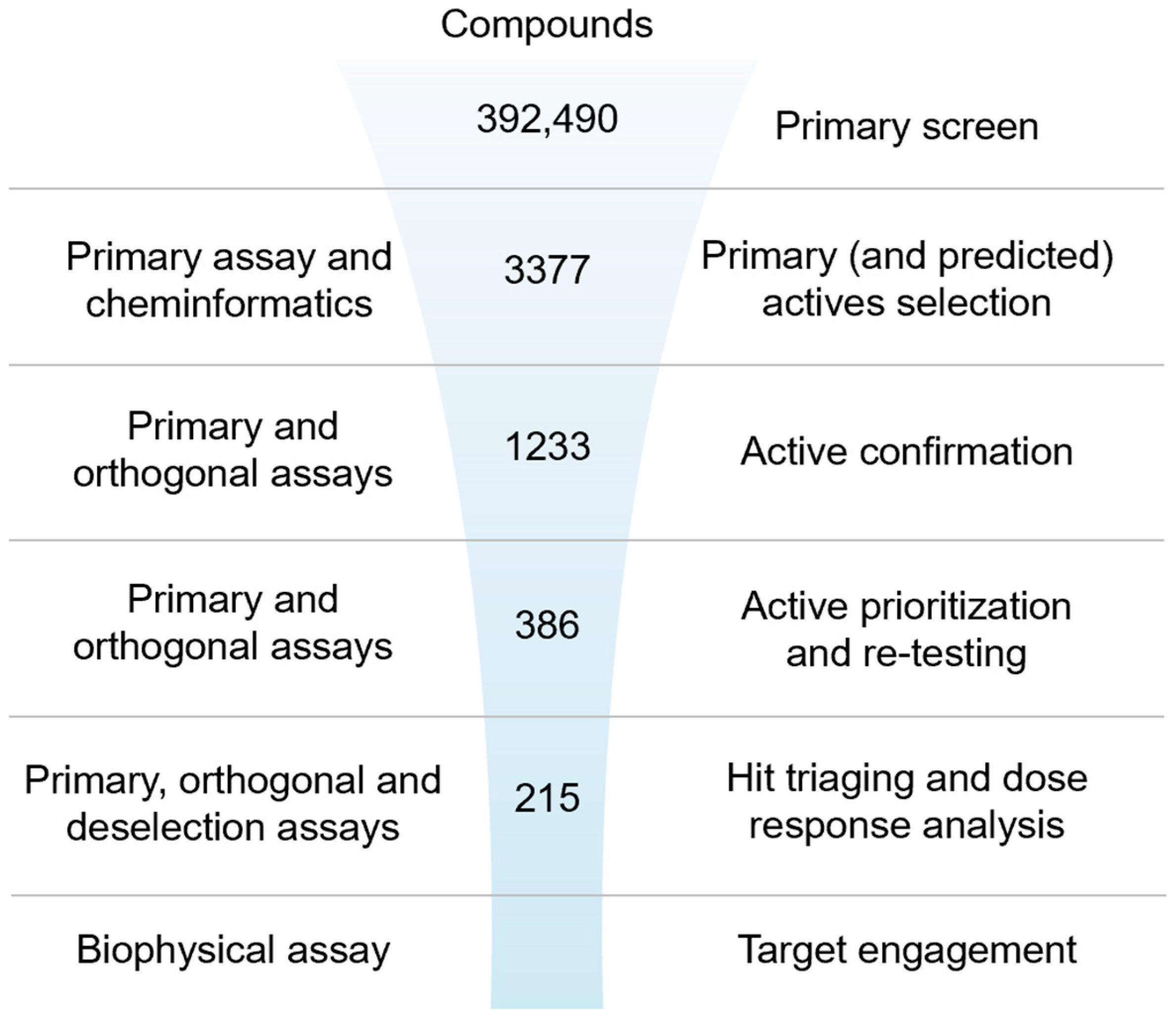
2.2. Screening Campaign and Hit Triaging
2.2.1. Primary Screen (Plate Statistics, Active Selection Criteria, Active Rate, NN, and Bayesian Active List Enrichment)
2.2.2. Active Confirmation in Primary and Orthogonal Assays (Plate Statistics, Active Selection Criteria, Confirmation Rate)
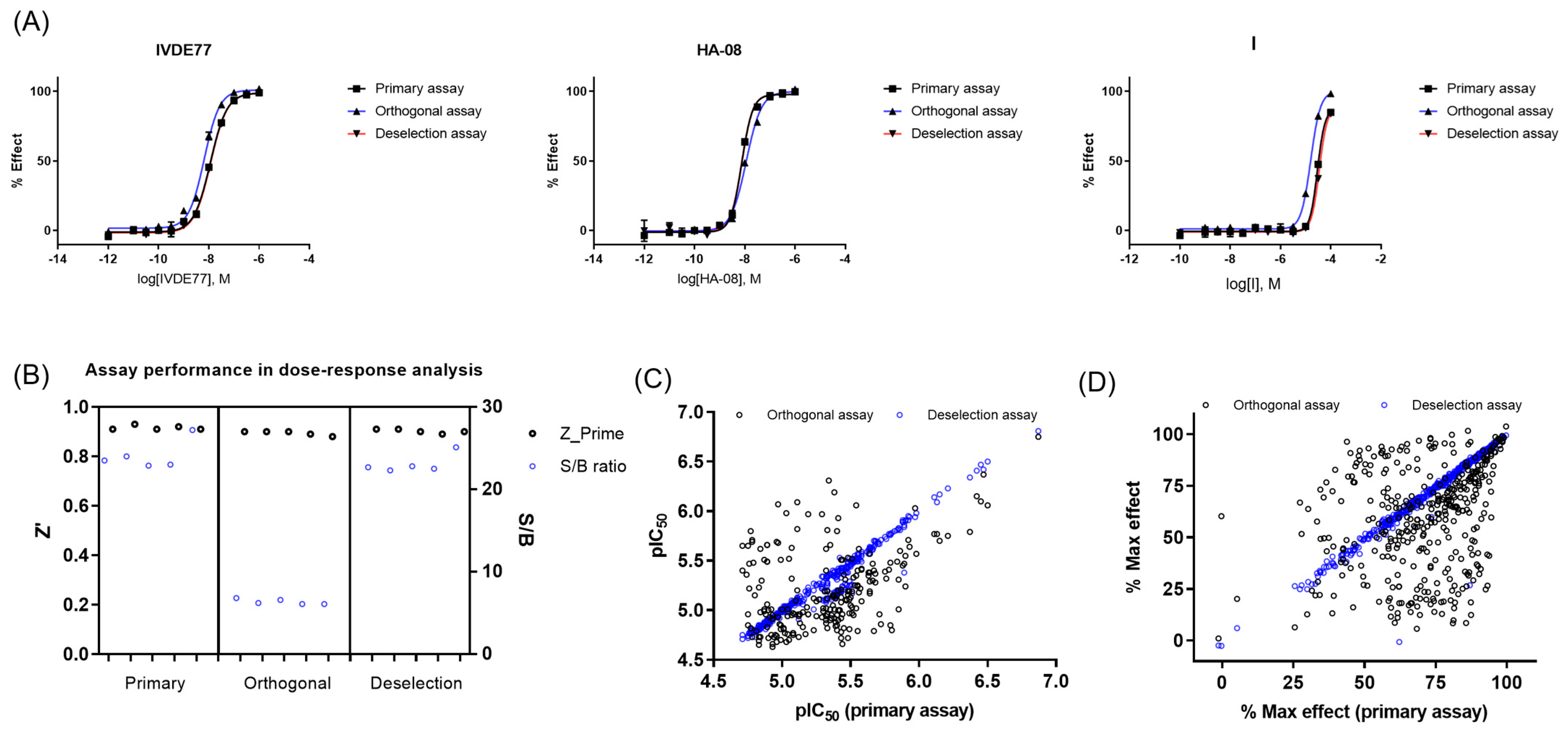
2.2.3. Dose–Response Curve (DRC) Analysis
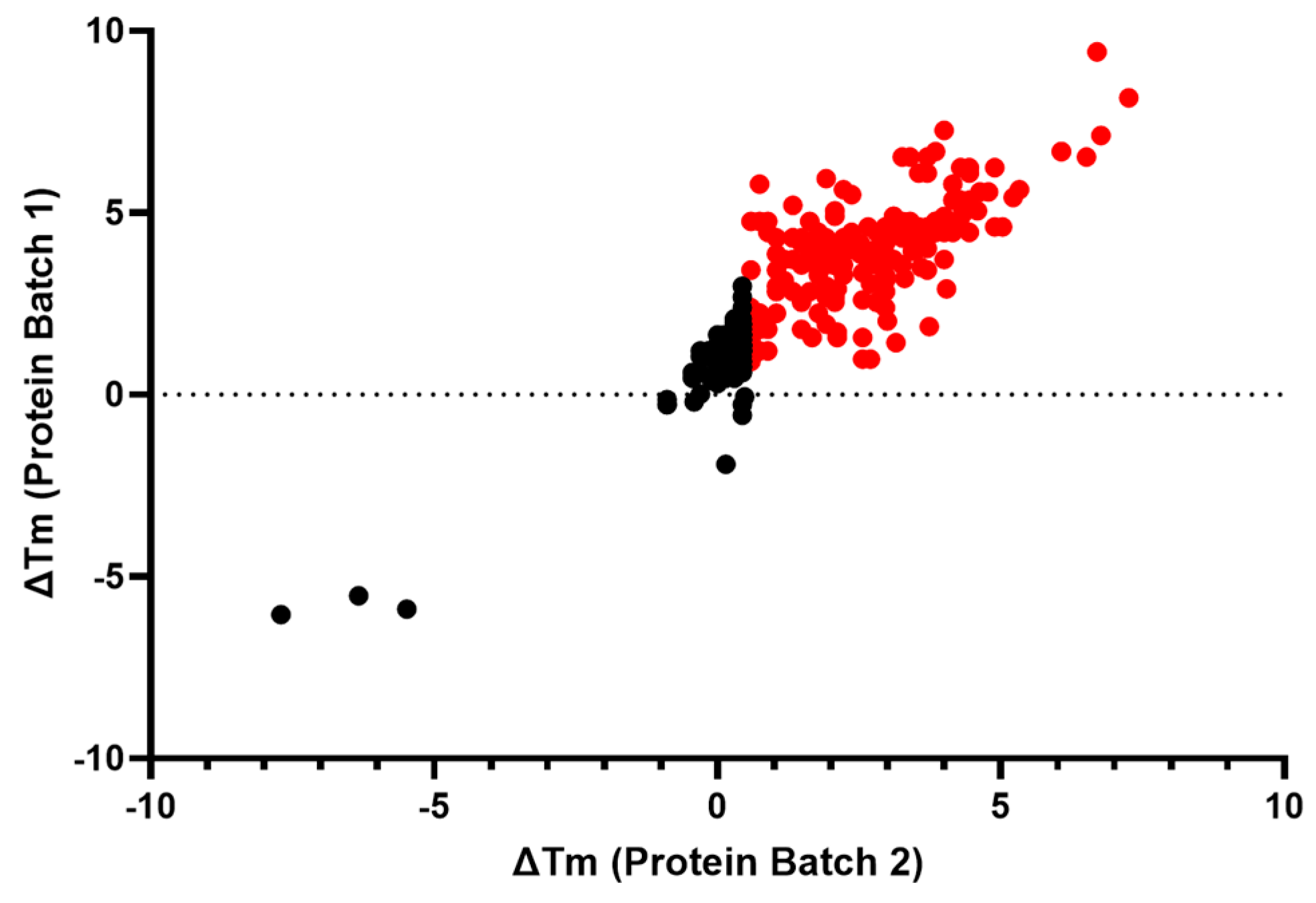
2.2.4. Biophysical Target Engagement Assay
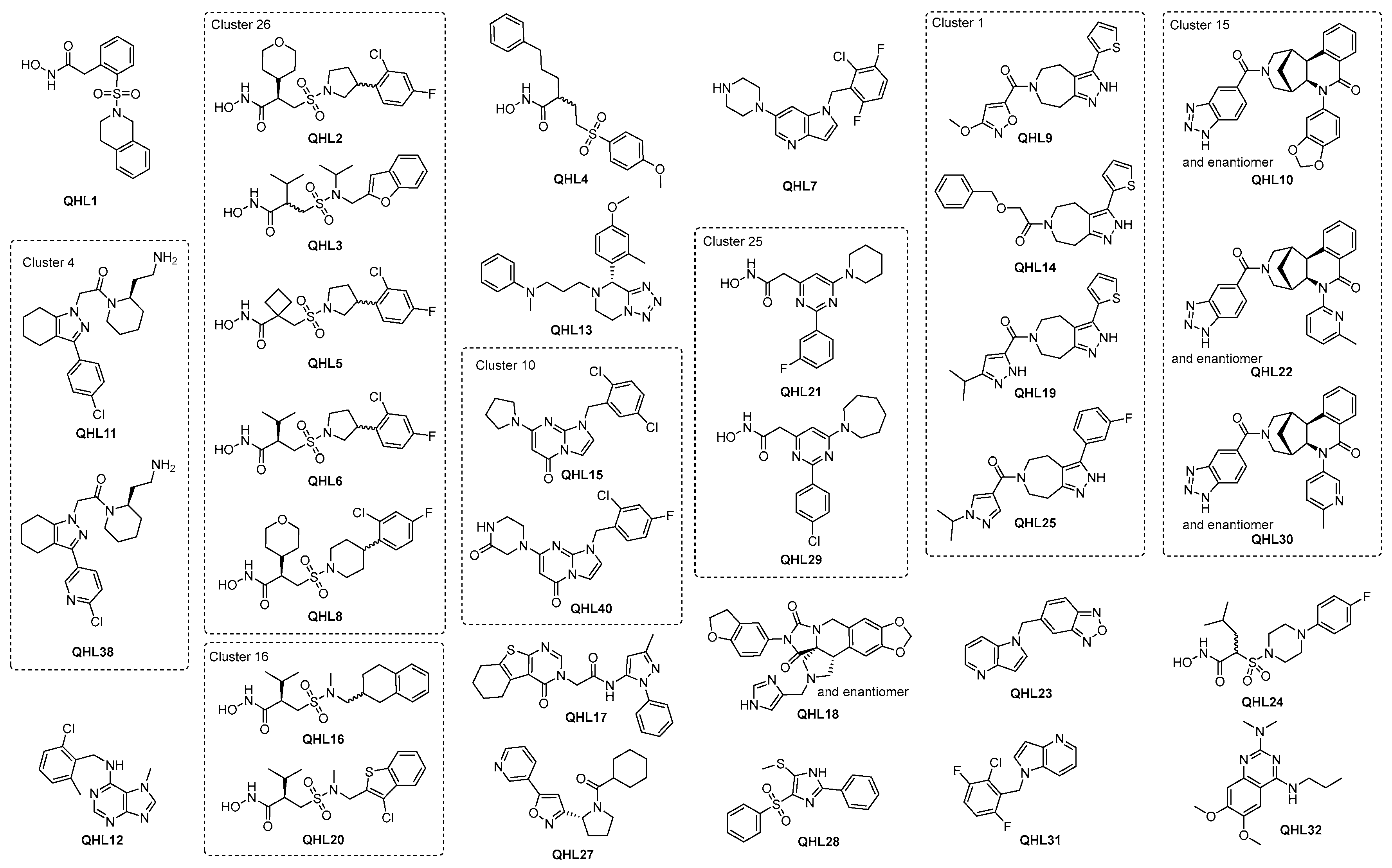
2.2.5. Hit Prioritization
2.3. Resynthesis of the Most Potent Hit
3. Discussion
4. Materials and Methods
4.1. Reference Compounds and Assay Substrates
4.2. Assay Buffers and Reagents
4.3. Assay Protocols
4.3.1. Primary Fluorescence IRAP Assay
4.3.2. Orthogonal Absorbance IRAP Assay
4.3.3. Deselection Fluorescence IRAP Assay
4.3.4. Thermal Shift (TSA) IRAP Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braszko, J.J.; Kupryszewski, G.; Witczuk, B.; Wiśniewski, K. Angiotensin Ii-(3–8)-Hexapeptide Affects Motor Activity, Performance of Passive Avoidance and a Conditioned Avoidance Response in Rats. Neuroscience 1988, 27, 777–783. [Google Scholar] [CrossRef]
- Wright, J.W.; Miller-Wing, A.V.; Shaffer, M.J.; Higginson, C.; Wright, D.E.; Hanesworth, J.M.; Harding, J.W. Angiotensin II(3–8) (ANG IV) Hippocampal Binding: Potential Role in the Facilitation of Memory. Brain Res. Bull. 1993, 32, 497–502. [Google Scholar] [CrossRef]
- Wright, J.W.; Clemens, J.A.; Panetta, J.A.; Smalstig, E.B.; Weatherly, L.A.S.; Kramár, E.A.; Pederson, E.S.; Mungall, B.H.; Harding, J.W. Effects of LY231617 and Angiotensin IV on Ischemia-Induced Deficits in Circular Water Maze and Passive Avoidance Performance in Rats. Brain Res. 1996, 717, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Mustafa, T.; McDowall, S.G.; Mendelsohn, F.A.O.; Brennan, M.; Lew, R.A.; Albiston, A.L.; Chai, S.Y. Structure-Activity Study of LVV-Hemorphin-7: Angiotensin AT4 Receptor Ligand and Inhibitor of Insulin-Regulated Aminopeptidase. J. Pharmacol. Exp. Ther. 2003, 305, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Culman, J.; Hörtnagl, H.; Zhao, Y.; Gerova, N.; Timm, M.; Blume, A.; Zimmermann, M.; Seidel, K.; Dirnagl, U.; et al. Angiotensin AT2 Receptor Protects against Cerebral Ischemia-Induced Neuronal Injury. FASEB J. 2005, 19, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Braszko, J.J.; Wielgat, P.; Walesiuk, A. Effect of D3 Dopamine Receptors Blockade on the Cognitive Effects of Angiotensin IV in Rats. Neuropeptides 2008, 42, 301–309. [Google Scholar] [CrossRef] [PubMed]
- De Bundel, D.; Smolders, I.; Yang, R.; Albiston, A.L.; Michotte, Y.; Chai, S.Y. Angiotensin IV and LVV-Haemorphin 7 Enhance Spatial Working Memory in Rats: Effects on Hippocampal Glucose Levels and Blood Flow. Neurobiol. Learn. Mem. 2009, 92, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Albiston, A.L.; Allen, A.M.; Mendelsohn, F.A.O.; Ping, S.E.; Barrett, G.L.; Murphy, M.; Morris, M.J.; Mcdowall, S.G.; Chai, S.Y. Effect of I.C.V. Injection of AT4 Receptor Ligands, NLE 1-Angiotensin IV and LVV-Hemorphin 7, on Spatial Learning in Rats. Neuroscience 2004, 124, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Swanson, G.N.; Hanesworth, J.M.; Sardinia, M.F.; Coleman, J.K.M.; Wright, J.W.; Hall, K.L.; Miller-Wing, A.V.; Stobb, J.W.; Cook, V.I.; Harding, E.C.; et al. Discovery of a Distinct Binding Site for Angiotensin II (3–8), a Putative Angiotensin IV Receptor. Regul. Pept. 1992, 40, 409–419. [Google Scholar] [CrossRef]
- Harding, J.W.; Cook, V.I.; Miller-Wing, A.V.; Hanesworth, J.M.; Sardinia, M.F.; Hall, K.L.; Stobb, J.W.; Swanson, G.N.; Coleman, J.K.M.; Wright, J.W.; et al. Identification of an AII(3–8) [AIV] Binding Site in Guinea Pig Hippocampus. Brain Res. 1992, 583, 340–343. [Google Scholar] [CrossRef]
- Gard, P.R. Cognitive-Enhancing Effects of Angiotensin IV. BMC Neurosci. 2008, 9, S15. [Google Scholar] [CrossRef]
- Albiston, A.L.; Diwakarla, S.; Fernando, R.N.; Mountford, S.J.; Yeatman, H.R.; Morgan, B.; Pham, V.; Holien, J.K.; Parker, M.W.; Thompson, P.E.; et al. Identification and Development of Specific Inhibitors for Insulin-Regulated Aminopeptidase as a New Class of Cognitive Enhancers. Br. J. Pharmacol. 2011, 164, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.K.; Nation, D.A. Cognitive Benefits of Angiotensin IV and Angiotensin-(1–7): A Systematic Review of Experimental Studies. Neurosci. Biobehav. Rev. 2018, 92, 209–225. [Google Scholar] [CrossRef]
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Harding, J.W. Contributions by the Brain Renin-Angiotensin System to Memory, Cognition, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Albiston, A.L.; McDowall, S.G.; Matsacos, D.; Sim, P.; Clune, E.; Mustafa, T.; Lee, J.; Mendelsohn, F.A.O.; Simpson, R.J.; Connolly, L.M.; et al. Evidence That the Angiotensin IV (AT4) Receptor Is the Enzyme Insulin-Regulated Aminopeptidase. J. Biol. Chem. 2001, 276, 48623–48626. [Google Scholar] [CrossRef]
- Fernando, R.N.; Larm, J.; Albiston, A.L.; Chai, S.Y. Distribution and Cellular Localization of Insulin-Regulated Aminopeptidase in the Rat Central Nervous System. J. Comp. Neurol. 2005, 487, 372–390. [Google Scholar] [CrossRef] [PubMed]
- Rogi, T.; Tsujimoto, M.; Nakazato, H.; Mizutani, S.; Tomoda, Y. Human Placental Leucine Aminopeptidase/Oxytocinase: A New Member of Type II Membrane-Spanning Zinc Metallopeptidase Family. J. Biol. Chem. 1996, 271, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Herbst, J.J.; Ross, S.A.; Scott, H.M.; Bobin, S.A.; Morris, N.J.; Lienhard, G.E.; Keller, S.R. Insulin Stimulates Cell Surface Aminopeptidase Activity toward Vasopressin in Adipocytes. Am. J. Physiol.-Endocrinol. Metab. 1997, 272, E600–E606. [Google Scholar] [CrossRef]
- Matsumoto, H.; Nagasaka, T.; Hattori, A.; Rogi, T.; Tsuruoka, N.; Mizutani, S.; Tsujimoto, M. Expression of Placental Leucine Aminopeptidase/Oxytocinase in Neuronal Cells and Its Action on Neuronal Peptides. Eur. J. Biochem. 2001, 268, 3259–3266. [Google Scholar] [CrossRef]
- Lew, R.A.; Mustafa, T.; Ye, S.; McDowall, S.G.; Chai, S.Y.; Albiston, A.L. Angiotensin AT4 Ligands Are Potent, Competitive Inhibitors of Insulin Regulated Aminopeptidase (IRAP). J. Neurochem. 2003, 86, 344–350. [Google Scholar] [CrossRef]
- Gulpinar, M.; Yegen, B. The Physiology of Learning and Memory: Role of Peptides and Stress. Curr. Protein Pept. Sci. 2005, 5, 457–473. [Google Scholar] [CrossRef]
- Wallis, M.G.; Lankford, M.F.; Keller, S.R. Vasopressin Is a Physiological Substrate for the Insulin-Regulated Aminopeptidase IRAP. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Rimmele, U.; Hediger, K.; Heinrichs, M.; Klaver, P. Oxytocin Makes a Face in Memory Familiar. J. Neurosci. 2009, 29, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Seyer, B.; Diwakarla, S.; Burns, P.; Hallberg, A.; Grönbladh, A.; Hallberg, M.; Chai, S.Y. Insulin-Regulated Aminopeptidase Inhibitor-Mediated Increases in Dendritic Spine Density Are Facilitated by Glucose Uptake. J. Neurochem. 2020, 153, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Hermans, S.J.; Ascher, D.B.; Hancock, N.C.; Holien, J.K.; Michell, B.J.; Chai, S.Y.; Morton, C.J.; Parker, M.W. Crystal Structure of Human Insulin-Regulated Aminopeptidase with Specificity for Cyclic Peptides. Protein Sci. 2015, 24, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Mpakali, A.; Saridakis, E.; Harlos, K.; Zhao, Y.; Kokkala, P.; Georgiadis, D.; Giastas, P.; Papakyriakou, A.; Stratikos, E. Ligand-Induced Conformational Change of Insulin-Regulated Aminopeptidase: Insights on Catalytic Mechanism and Active Site Plasticity. J. Med. Chem. 2017, 60, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Mpakali, A.; Saridakis, E.; Giastas, P.; Maben, Z.; Stern, L.J.; Larhed, M.; Hallberg, M.; Stratikos, E. Structural Basis of Inhibition of Insulin-Regulated Aminopeptidase by a Macrocyclic Peptidic Inhibitor. ACS Med. Chem. Lett. 2020, 11, 1429–1434. [Google Scholar] [CrossRef]
- Vear, A.; Gaspari, T.; Thompson, P.; Chai, S.Y. Is There an Interplay Between the Functional Domains of IRAP? Front. Cell Dev. Biol. 2020, 8, 585237. [Google Scholar] [CrossRef]
- Chai, S.Y.; Yeatman, H.R.; Parker, M.W.; Ascher, D.B.; Thompson, P.E.; Mulvey, H.T.; Albiston, A.L. Development of Cognitive Enhancers Based on Inhibition of Insulin-Regulated Aminopeptidase. BMC Neurosci. 2008, 9, S14. [Google Scholar] [CrossRef]
- Hallberg, M. Targeting the Insulin-Regulated Aminopeptidase/AT4 Receptor for Cognitive Disorders. Drug News Perspect. 2009, 22, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Saveanu, L.; Carroll, O.; Weimershaus, M.; Guermonprez, P.; Firat, E.; Lindo, V.; Greer, F.; Davoust, J.; Kratzer, R.; Keller, S.R.; et al. IRAP Identifies an Endosomal Compartment Required for MHC Class i Cross-Presentation. Science 2009, 325, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Evnouchidou, I.; Chappert, P.; Benadda, S.; Zucchetti, A.; Weimershaus, M.; Bens, M.; Caillens, V.; Koumantou, D.; Lotersztajn, S.; van Endert, P.; et al. IRAP-Dependent Endosomal T Cell Receptor Signalling Is Essential for T Cell Responses. Nat. Commun. 2020, 11, 2779. [Google Scholar] [CrossRef]
- Telianidis, J.; Hunter, A.; Widdop, R.; Kemp-Harper, B.; Pham, V.; McCarthy, C.; Chai, S.Y. Inhibition of Insulin-Regulated Aminopeptidase Confers Neuroprotection in a Conscious Model of Ischemic Stroke. Sci. Rep. 2023, 13, 19722. [Google Scholar] [CrossRef] [PubMed]
- Axén, A.; Lindeberg, G.; Demaegdt, H.; Vauquelin, G.; Karlén, A.; Hallberg, M. Cyclic Insulin-Regulated Aminopeptidase (IRAP)/AT4 Receptor Ligands. J. Pept. Sci. 2006, 12, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Lukaszuk, A.; Demaegdt, H.; Szemenyei, E.; Tóth, G.; Tymecka, D.; Misicka, A.; Karoyan, P.; Vanderheyden, P.; Vauquelin, G.; Tourwé, D. β-Homo-Amino Acid Scan of Angiotensin IV. J. Med. Chem. 2008, 51, 2291–2296. [Google Scholar] [CrossRef] [PubMed]
- Lukaszuk, A.; Demaegdt, H.; Feytens, D.; Vanderheyden, P.; Vauquelin, G.; Tourwé, D. The Replacement of His(4) in Angiotensin IV by Conformationally Constrained Residues Provides Highly Potent and Selective Analogues. J. Med. Chem. 2009, 52, 5612–5618. [Google Scholar] [CrossRef]
- Nikolaou, A.; Van Den Eynde, I.; Tourwé, D.; Vauquelin, G.; Tóth, G.; Mallareddy, J.R.; Poglitsch, M.; Van Ginderachter, J.A.; Vanderheyden, P.M.L. [3H]IVDE77, a Novel Radioligand with High Affinity and Selectivity for the Insulin-Regulated Aminopeptidase. Eur. J. Pharmacol. 2013, 702, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Demaegdt, H.; Vauquelin, G.; Lindeberg, G.; Karlén, A.; Hallberg, M.; Erdélyi, M.; Hallberg, A. Disulfide Cyclized Tripeptide Analogues of Angiotensin IV as Potent and Selective Inhibitors of Insulin-Regulated Aminopeptidase (IRAP). J. Med. Chem. 2010, 53, 8059–8071. [Google Scholar] [CrossRef]
- Diwakarla, S.; Nylander, E.; Grönbladh, A.; Vanga, S.R.; Khan, Y.S.; Gutiérrez-de-Terán, H.; Ng, L.; Pham, V.; Sävmarker, J.; Lundbäck, T.; et al. Binding to and Inhibition of Insulin-Regulated Aminopeptidase by Macrocyclic Disulfides Enhances Spine Density. Mol. Pharmacol. 2016, 89, 413–424. [Google Scholar] [CrossRef]
- Stam, F.; Florén Lind, S.; Schroff, A.; Zelleroth, S.; Nylander, E.; Gising, J.; Grönbladh, A.; Larhed, M.; Hallberg, M. Hydrogen Peroxide Induced Toxicity Is Reversed by the Macrocyclic IRAP-Inhibitor HA08 in Primary Hippocampal Cell Cultures. Curr. Issues Mol. Biol. 2022, 44, 5000–5012. [Google Scholar] [CrossRef] [PubMed]
- Albiston, A.L.; Morton, C.J.; Ng, H.L.; Pham, V.; Yeatman, H.R.; Ye, S.; Fernando, R.N.; De Bundel, D.; Ascher, D.B.; Mendelsohn, F.A.O.; et al. Identification and Characterization of a New Cognitive Enhancer Based on Inhibition of Insulin-regulated Aminopeptidase. FASEB J. 2008, 22, 4209–4217. [Google Scholar] [CrossRef] [PubMed]
- Thunnissen, M.M.G.M.; Nordlund, P.; Haeggström, J.Z. Crystal Structure of Human Leukotriene A4 Hydrolase, a Bifunctional Enzyme in Inflammation. Nat. Struct. Biol. 2001, 8, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Engen, K.; Rosenström, U.; Axelsson, H.; Konda, V.; Dahllund, L.; Otrocka, M.; Sigmundsson, K.; Nikolaou, A.; Vauquelin, G.; Hallberg, M.; et al. Identification of Drug-Like Inhibitors of Insulin-Regulated Aminopeptidase Through Small-Molecule Screening. Assay Drug Dev. Technol. 2016, 14, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Borhade, S.R.; Rosenström, U.; Sävmarker, J.; Lundbäck, T.; Jenmalm-Jensen, A.; Sigmundsson, K.; Axelsson, H.; Svensson, F.; Konda, V.; Sköld, C.; et al. Inhibition of Insulin-Regulated Aminopeptidase (IRAP) by Arylsulfonamides. ChemistryOpen 2014, 3, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Diwakarla, S.; Nylander, E.; Grönbladh, A.; Vanga, S.R.; Khan, Y.S.; Gutiérrez-De-Terán, H.; Sävmarker, J.; Ng, L.; Pham, V.; Lundbäck, T.; et al. Aryl Sulfonamide Inhibitors of Insulin-Regulated Aminopeptidase Enhance Spine Density in Primary Hippocampal Neuron Cultures. ACS Chem. Neurosci. 2016, 7, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Engen, K.; Vanga, S.R.; Lundbäck, T.; Agalo, F.; Konda, V.; Jensen, A.J.; Åqvist, J.; Gutiérrez-de-Terán, H.; Hallberg, M.; Larhed, M.; et al. Synthesis, Evaluation and Proposed Binding Pose of Substituted Spiro-Oxindole Dihydroquinazolinones as IRAP Inhibitors. ChemistryOpen 2020, 9, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Engen, K.; Lundbäck, T.; Yadav, A.; Puthiyaparambath, S.; Rosenström, U.; Gising, J.; Jenmalm-jensen, A.; Hallberg, M.; Larhed, M. Inhibition of Insulin-Regulated Aminopeptidase by Imidazo [1,5-α] Pyridines—Synthesis and Evaluation. Int. J. Mol. Sci. 2024, 25, 2516. [Google Scholar] [CrossRef] [PubMed]
- Temponeras, I.; Chiniadis, L.; Papakyriakou, A.; Stratikos, E. Discovery of Selective Inhibitor Leads by Targeting an Allosteric Site in Insulin-Regulated Aminopeptidase. Pharmaceuticals 2021, 14, 584. [Google Scholar] [CrossRef]
- Medve, L.; Gealageas, R.; Lam, B.V.; Guillaume, V.; Castillo-Aguilera, O.; Camberlein, V.; Piveteau, C.; Rosell, M.; Fleau, C.; Warenghem, S.; et al. Modulators of HERAP2 Discovered by High-Throughput Screening. Eur. J. Med. Chem. 2021, 211, 113053. [Google Scholar] [CrossRef]
- Mpakali, A.; Barla, I.; Lu, L.; Ramesh, K.M.; Thomaidis, N.; Stern, L.J.; Giastas, P.; Stratikos, E. Mechanisms of Allosteric Inhibition of Insulin-Regulated Aminopeptidase. J. Mol. Biol. 2024, 436, 168449. [Google Scholar] [CrossRef] [PubMed]
- Papakyriakou, A.; Zervoudi, E.; Tsoukalidou, S.; Mauvais, F.-X.; Sfyroera, G.; Mastellos, D.C.; van Endert, P.; Theodorakis, E.A.; Vourloumis, D.; Stratikos, E. 3,4-Diaminobenzoic Acid Derivatives as Inhibitors of the Oxytocinase Subfamily of M1 Aminopeptidases with Immune-Regulating Properties. J. Med. Chem. 2015, 58, 1524–1543. [Google Scholar] [CrossRef] [PubMed]
- Kokkala, P.; Mpakali, A.; Mauvais, F.-X.; Papakyriakou, A.; Daskalaki, I.; Petropoulou, I.; Kavvalou, S.; Papathanasopoulou, M.; Agrotis, S.; Fonsou, T.-M.; et al. Optimization and Structure–Activity Relationships of Phosphinic Pseudotripeptide Inhibitors of Aminopeptidases That Generate Antigenic Peptides. J. Med. Chem. 2016, 59, 9107–9123. [Google Scholar] [CrossRef]
- Vourloumis, D.; Mavridis, I.; Athanasoulis, A.; Temponeras, I.; Koumantou, D.; Giastas, P.; Mpakali, A.; Magrioti, V.; Leib, J.; van Endert, P.; et al. Discovery of Selective Nanomolar Inhibitors for Insulin-Regulated Aminopeptidase Based on α-Hydroxy-β-Amino Acid Derivatives of Bestatin. J. Med. Chem. 2022, 65, 10098–10117. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, D.; Ziotopoulou, A.; Kaloumenou, E.; Lelis, A.; Papasava, A. The Discovery of Insulin-Regulated Aminopeptidase (IRAP) Inhibitors: A Literature Review. Front. Pharmacol. 2020, 11, 585838. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, M.; Larhed, M. From Angiotensin IV to Small Peptidemimetics Inhibiting Insulin-Regulated Aminopeptidase. Front. Pharmacol. 2020, 11, 590855. [Google Scholar] [CrossRef]
- Barlow, N.; Thompson, P.E. IRAP Inhibitors: M1-Aminopeptidase Family Inspiration. Front. Pharmacol. 2020, 11, 585930. [Google Scholar] [CrossRef]
- Brown, D.G. An Analysis of Successful Hit-to-Clinical Candidate Pairs. J. Med. Chem. 2023, 66, 7101–7139. [Google Scholar] [CrossRef]
- Jones, P.S.; Boucharens, S.; McElroy, S.P.; Morrison, A.; Honarnejad, S.; van Boeckel, S.; van den Hurk, H.; Basting, D.; Hüser, J.; Jaroch, S.; et al. IMI European Lead Factory—Democratizing Access to High-Throughput Screening. Nat. Rev. Drug Discov. 2021, 21, 245–246. [Google Scholar] [CrossRef]
- van Vlijmen, H.; Ortholand, J.Y.; Li, V.M.J.; de Vlieger, J.S.B. The European Lead Factory: An Updated HTS Compound Library for Innovative Drug Discovery. Drug Discov. Today 2021, 26, 2406–2413. [Google Scholar] [CrossRef]
- Honarnejad, S.; van Boeckel, S.; van den Hurk, H.; van Helden, S. Hit Discovery for Public Target Programs in the European Lead Factory: Experiences and Output from Assay Development and Ultra-High-Throughput Screening. SLAS Discov. 2021, 26, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. SLAS Discov. 2001, 6, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Mcelroy, S.; Morrison, A.; Pannifer, A. The Importance of Triaging in Determining the Quality of Output from High-Throughput Screening. Future Med. Chem. 2015, 7, 1847–1852. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Besnard, J.; Jones, P.S.; Hopkins, A.L.; Pannifer, A.D. The Joint European Compound Library: Boosting Precompetitive Research. Drug Discov. Today 2015, 20, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Buker, S.M.; Boriack-Sjodin, P.A.; Copeland, R.A. Enzyme–Inhibitor Interactions and a Simple, Rapid Method for Determining Inhibition Modality. SLAS Discov. 2019, 24, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Llowarch, P.; Usselmann, L.; Ivanov, D.; Holdgate, G.A. Thermal Unfolding Methods in Drug Discovery. Biophys. Rev. 2023, 4, 021305. [Google Scholar] [CrossRef] [PubMed]
- Wielens, J.; Headey, S.J.; Rhodes, D.I.; Mulder, R.J.; Dolezal, O.; Deadman, J.J.; Newman, J.; Chalmers, D.K.; Parker, M.W.; Peat, T.S.; et al. Parallel Screening of Low Molecular Weight Fragment Libraries: Do Differences in Methodology Affect Hit Identification? J. Biomol. Screen. 2013, 18, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Schiebel, J.; Radeva, N.; Köster, H.; Metz, A.; Krotzky, T.; Kuhnert, M.; Diederich, W.E.; Heine, A.; Neumann, L.; Atmanene, C.; et al. One Question, Multiple Answers: Biochemical and Biophysical Screening Methods Retrieve Deviating Fragment Hit Lists. ChemMedChem 2015, 10, 1511–1521. [Google Scholar] [CrossRef]
- Schiebel, J.; Radeva, N.; Krimmer, S.G.; Wang, X.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Metz, A.; Huschmann, F.U.; Weiss, M.S.; et al. Six Biophysical Screening Methods Miss a Large Proportion of Crystallographically Discovered Fragment Hits: A Case Study. ACS Chem. Biol. 2016, 11, 1693–1701. [Google Scholar] [CrossRef]
- Day, J.A.; Cohen, S.M. Investigating the Selectivity of Metalloenzyme Inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; Macdonald, S.J.F. How Drug-like Are “ugly” Drugs: Do Drug-Likeness Metrics Predict ADME Behaviour in Humans? Drug Discov. Today 2014, 19, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Demaegdt, H.; Vauquelin, G.; Lindeberg, G.; Karlén, A.; Hallberg, M. Ligands to the (IRAP)/AT4 receptor encompassing a 4-hydroxydiphenylmethane scaffold replacing Tyr2. Bioorg. Med. Chem. 2008, 16, 6924–6935. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QHL | Cluster | Fluorescence IC50 (µM) | Fluorescence pIC50 | Absorbance pIC50 | Fluorescence + Zn pIC50 | TSA (∆°C) | Mw (g/mol) | HBA/HBD | TPSA (Å2) | LogD | LipE | QED |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NA | 0.32 | 6.50 | 6.06 | 6.50 | 6.77 | 346.40 | 6/2 | 95.09 | 1.65 | 4.85 | 0.65 |
| 2 | 26 | 0.35 | 6.45 | 6.10 | 6.47 | 3.70 | 434.91 | 7/2 | 104.32 | 1.56 | 4.89 | 0.52 |
| 3 | 26 | 0.38 | 6.42 | 6.15 | 6.41 | 5.22 | 382.47 | 7/2 | 108.22 | 2.34 | 4.08 | 0.54 |
| 4 | NA | 0.71 | 6.15 | 5.70 | 6.17 | 4.00 | 391.48 | 6/2 | 101.08 | 3.64 | 2.51 | 0.48 |
| 5 | 26 | 1.20 | 5.92 | 5.71 | 5.95 | 4.29 | 390.86 | 6/2 | 95.09 | 2.13 | 3.79 | 0.59 |
| 6 | 26 | 1.26 | 5.90 | 5.24 | 5.91 | 3.55 | 392.87 | 6/2 | 95.09 | 2.33 | 3.57 | 0.57 |
| 7 | NA | 1.29 | 5.89 | <4.70 | 5.86 | 0.30 | 362.80 | 4/1 | 33.09 | 3.79 | 2.10 | 0.72 |
| 8 | 26 | 1.32 | 5.88 | 5.45 | 5.89 | 4.61 | 448.94 | 7/2 | 104.32 | 1.94 | 3.94 | 0.51 |
| 9 | 1 | 1.51 | 5.82 | <4.70 | 5.80 | 0.15 | 344.39 | 7/1 | 112.49 | 2.84 | 2.98 | 0.79 |
| 10 | 15 | 1.58 | 5.80 | 4.80 | 5.78 | 0.30 | 493.51 | 9/1 | 100.65 | 3.27 | 2.53 | 0.46 |
| 11 | 4 | 1.66 | 5.78 | 5.41 | 5.77 | 1.92 | 400.94 | 5/2 | 64.15 | 3.21 | 2.57 | 0.83 |
| 12 | NA | 1.91 | 5.72 | 4.86 | 5.71 | −0.15 | 287.75 | 5/1 | 55.63 | 4.04 | 1.68 | 0.80 |
| 13 | NA | 2.14 | 5.67 | 4.79 | 5.70 | −1.91 | 392.50 | 7/0 | 59.31 | 3.28 | 2.39 | 0.61 |
| 14 | 1 | 2.24 | 5.65 | <4.70 | 5.63 | 0.15 | 367.46 | 5/1 | 86.46 | 3.23 | 2.42 | 0.75 |
| 15 | 10 | 2.40 | 5.62 | <4.70 | 5.59 | −0.89 | 363.24 | 5/0 | 39.15 | 2.08 | 3.54 | 0.83 |
| 16 | 16 | 2.63 | 5.58 | 5.42 | 5.55 | 2.07 | 368.49 | 6/2 | 95.09 | 2.44 | 3.14 | 0.57 |
| 17 | NA | 2.63 | 5.58 | <4.70 | 5.49 | 0.30 | 419.50 | 7/1 | 107.82 | 3.39 | 2.19 | 0.70 |
| 18 | NA | 2.82 | 5.55 | 5.25 | 5.57 | 1.92 | 485.49 | 10/1 | 100.23 | 2.53 | 3.02 | 0.56 |
| 19 | 1 | 2.82 | 5.55 | 4.79 | 5.53 | 0.30 | 355.46 | 6/2 | 105.91 | 3.72 | 1.83 | 0.75 |
| 20 | 16 | 2.82 | 5.55 | 5.24 | 5.5 | 4.15 | 404.93 | 6/2 | 123.33 | 2.84 | 2.71 | 0.54 |
| 21 | 25 | 2.95 | 5.53 | 5.25 | 5.55 | 2.66 | 330.36 | 6/2 | 78.35 | 2.96 | 2.57 | 0.67 |
| 22 | 15 | 3.09 | 5.51 | <4.70 | 5.52 | 0.15 | 464.52 | 8/1 | 95.08 | 3.17 | 2.34 | 0.49 |
| 23 | NA | 3.09 | 5.51 | <4.70 | 5.49 | 0.44 | 250.26 | 5/0 | 56.74 | 2.90 | 2.61 | 0.55 |
| 24 | NA | 3.09 | 5.51 | 5.52 | 5.26 | 4.00 | 387.47 | 7/2 | 98.33 | 1.94 | 3.57 | 0.55 |
| 25 | 1 | 3.16 | 5.50 | <4.70 | 5.46 | 0.00 | 367.42 | 6/1 | 66.81 | 3.20 | 2.30 | 0.77 |
| 26 | NA | 3.24 | 5.49 | 5.11 | 5.45 | 4.44 | 428.52 | 7/2 | 104.32 | 2.58 | 2.91 | 0.51 |
| 27 | NA | 3.24 | 5.49 | 4.97 | 5.29 | 1.48 | 325.40 | 5/0 | 59.23 | 3.35 | 2.14 | 0.87 |
| 28 | NA | 3.24 | 5.49 | 4.81 | 5.25 | 0.15 | 330.42 | 4/1 | 96.50 | 3.95 | 1.54 | 0.73 |
| 29 | 25 | 3.31 | 5.48 | 5.53 | 5.49 | 1.92 | 360.84 | 6/2 | 78.35 | 3.88 | 1.60 | 0.64 |
| 30 | 15 | 3.47 | 5.46 | <4.70 | 5.41 | 0.30 | 464.52 | 8/1 | 95.08 | 3.03 | 2.43 | 0.49 |
| 31 | NA | 3.47 | 5.46 | <4.70 | 5.38 | 0.00 | 278.68 | 2/0 | 17.82 | 4.41 | 1.05 | 0.64 |
| 32 | NA | 3.47 | 5.46 | <4.70 | 5.21 | 0.15 | 290.36 | 6/1 | 59.51 | 2.36 | 3.10 | 0.88 |
| 38 | 4 | 4.68 | 5.33 | 4.96 | 5.34 | 2.22 | 401.93 | 6/2 | 77.04 | 2.27 | 3.06 | 0.78 |
| 40 | 10 | 4.68 | 5.33 | <4.70 | 5.28 | 0.30 | 375.78 | 7/1 | 68.25 | 0.95 | 4.38 | 0.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gising, J.; Honarnejad, S.; Bras, M.; Baillie, G.L.; McElroy, S.P.; Jones, P.S.; Morrison, A.; Beveridge, J.; Hallberg, M.; Larhed, M. The Discovery of New Inhibitors of Insulin-Regulated Aminopeptidase by a High-Throughput Screening of 400,000 Drug-like Compounds. Int. J. Mol. Sci. 2024, 25, 4084. https://doi.org/10.3390/ijms25074084
Gising J, Honarnejad S, Bras M, Baillie GL, McElroy SP, Jones PS, Morrison A, Beveridge J, Hallberg M, Larhed M. The Discovery of New Inhibitors of Insulin-Regulated Aminopeptidase by a High-Throughput Screening of 400,000 Drug-like Compounds. International Journal of Molecular Sciences. 2024; 25(7):4084. https://doi.org/10.3390/ijms25074084
Chicago/Turabian StyleGising, Johan, Saman Honarnejad, Maaike Bras, Gemma L. Baillie, Stuart P. McElroy, Philip S. Jones, Angus Morrison, Julia Beveridge, Mathias Hallberg, and Mats Larhed. 2024. "The Discovery of New Inhibitors of Insulin-Regulated Aminopeptidase by a High-Throughput Screening of 400,000 Drug-like Compounds" International Journal of Molecular Sciences 25, no. 7: 4084. https://doi.org/10.3390/ijms25074084
APA StyleGising, J., Honarnejad, S., Bras, M., Baillie, G. L., McElroy, S. P., Jones, P. S., Morrison, A., Beveridge, J., Hallberg, M., & Larhed, M. (2024). The Discovery of New Inhibitors of Insulin-Regulated Aminopeptidase by a High-Throughput Screening of 400,000 Drug-like Compounds. International Journal of Molecular Sciences, 25(7), 4084. https://doi.org/10.3390/ijms25074084