
Neuroinflammation and Epilepsy: From Pathophysiology to Therapies Based on Repurposing Drugs


Abstract
:1. Introduction
Neuroinflammation: A General Overview
2. Methodology
3. Cell Types of the CNS Involved in Neuroinflammation
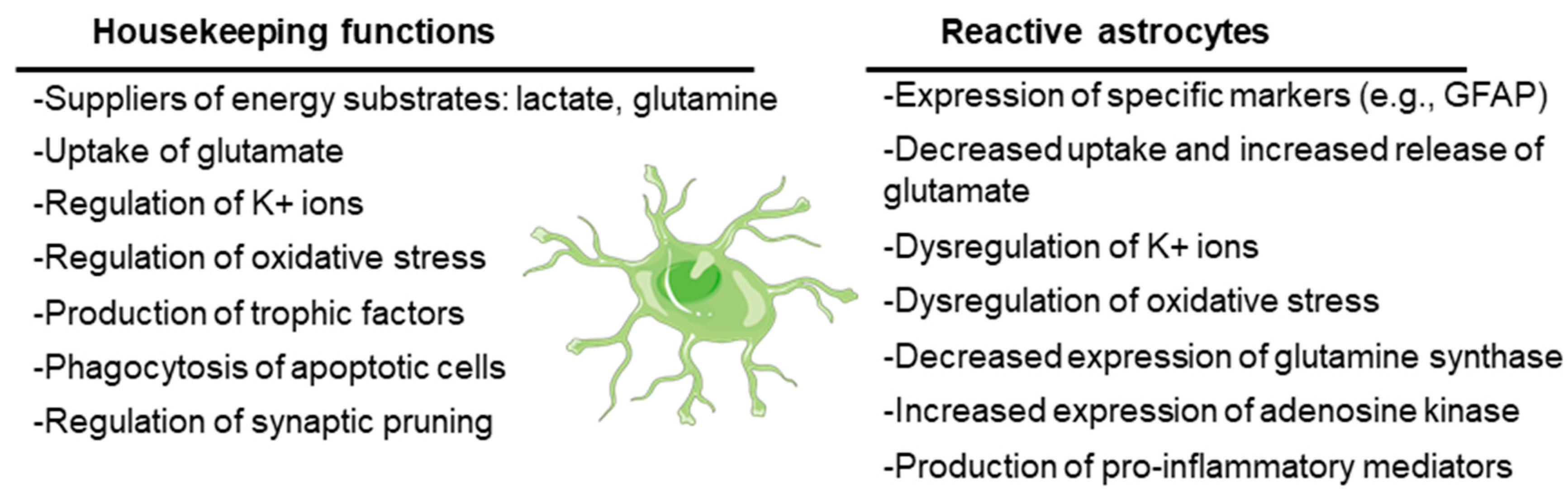
3.1. Astrocytes
3.2. Microglia
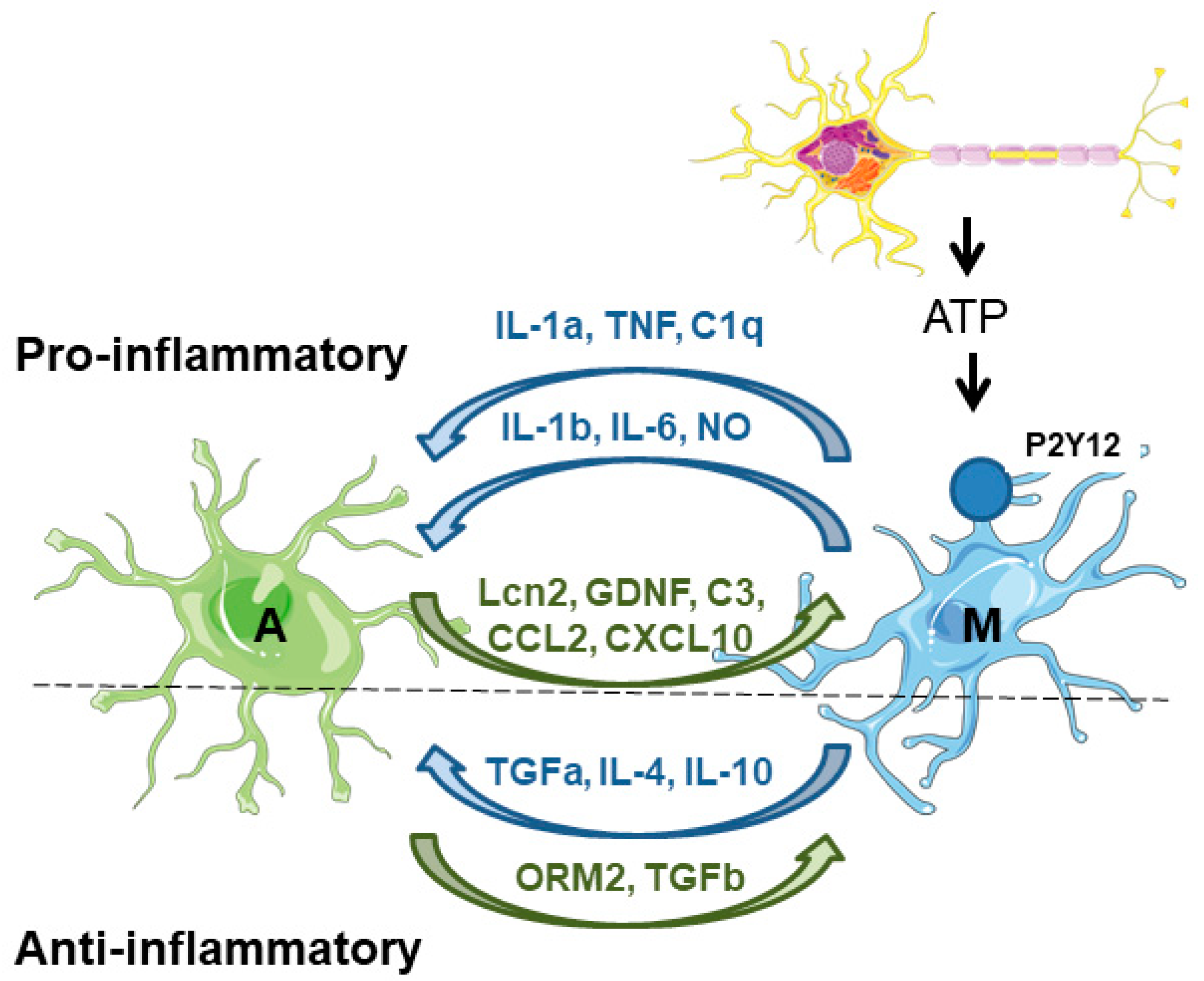
3.3. Microglia–Astrocyte Crosstalk
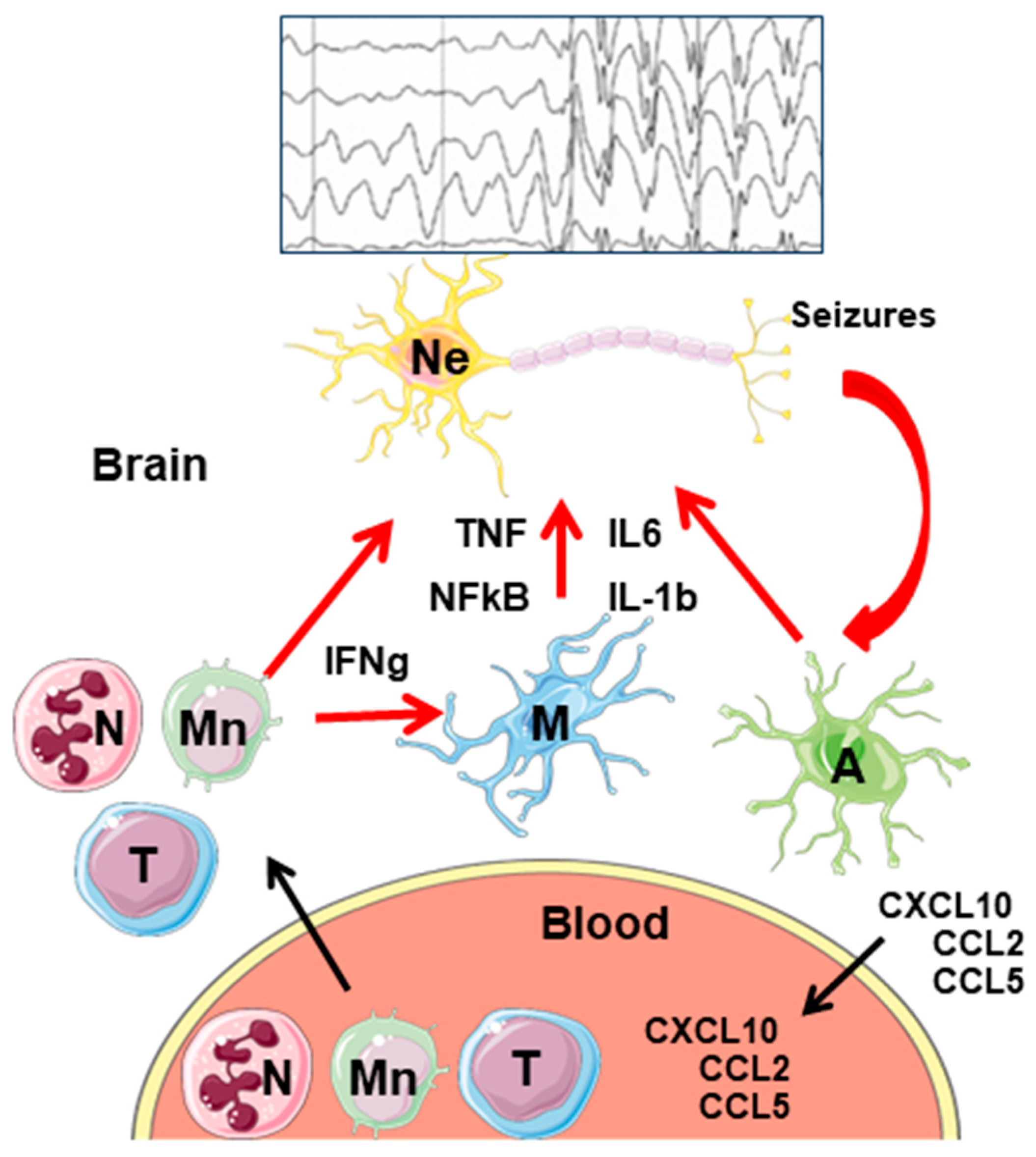
3.4. Peripheral Immune Cells Infiltration into the CNS
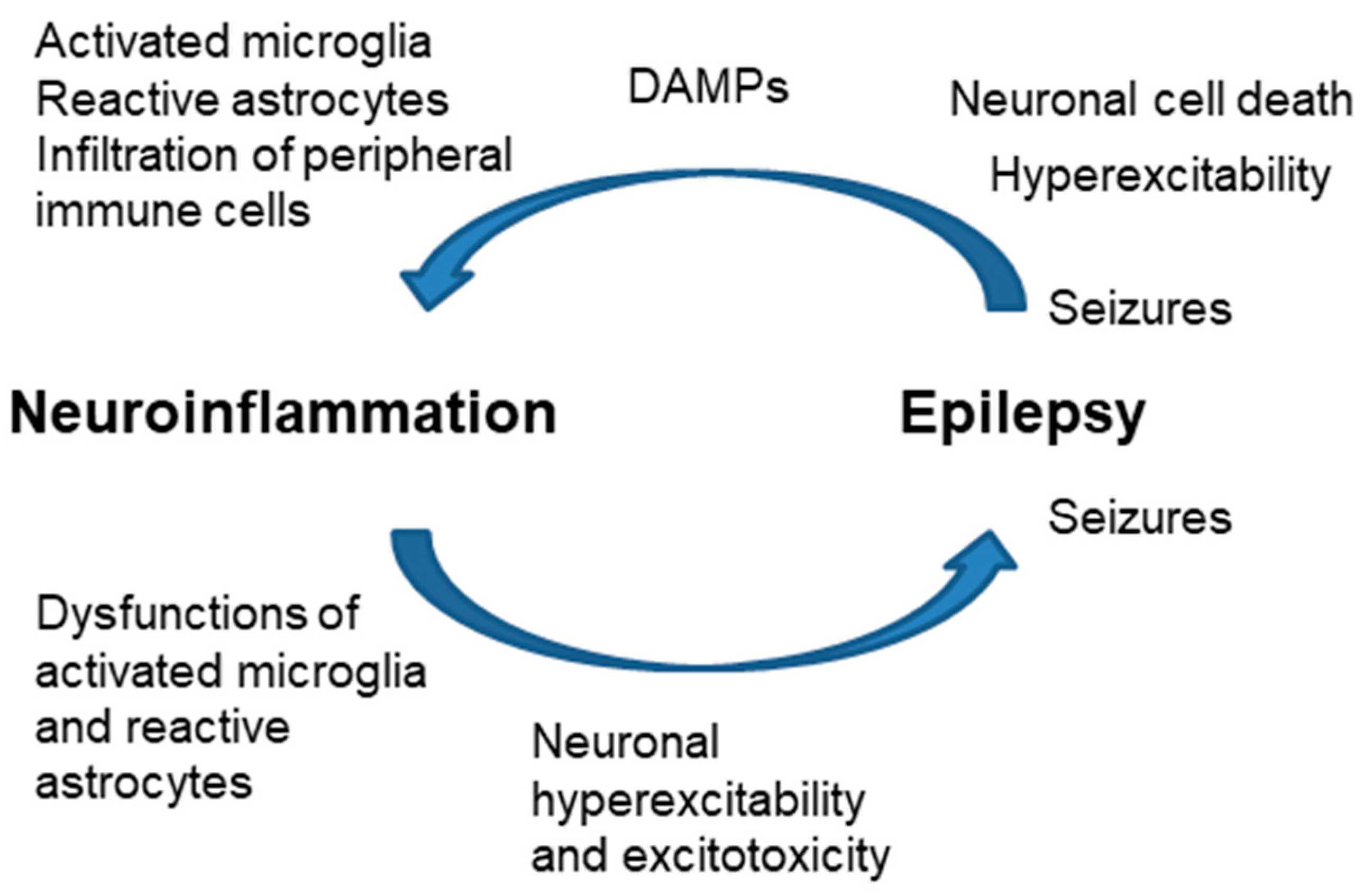
4. Neuroinflammation and Epilepsy
5. Use of Repurposing Drugs as an Anti-Inflammatory Therapeutic Approach in Epilepsy
5.1. Metformin
5.2. Fingolimod
5.3. Dimethyl Fumarate
5.4. Propranolol

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Primary Indication | Primary Mechanism of Action | Secondary Indication | Secondary Mechanism of Action | References of Secondary Indication |
|---|---|---|---|---|---|
| Metformin | Type 2 Diabetes | Reduces hepatic glucose production | Anti-seizure effect | Anti-inflammatory, with AMPK-dependent and -independent effects | Animal models: [75,76,77,78] Humans: [79] |
| Fingolimod | Multiple sclerosis | Antagonist of sphingosine-1-phosphate receptors with broad action at the central nervous system | Anti-seizure effect | Anti-inflammatory; prevents leukocyte infiltration into the brain parenchyma | Animal models: [83,84,85,86,87] Humans: [88] |
| Dimethyl Fumarate | Multiple sclerosis | Antioxidant; improves erythroid 2-related factor-dependent action | Anti-seizure effect | Anti-inflammatory; prevents leukocyte infiltration into the brain parenchyma | Animal models: [94,95,96] |
| Propranolol | b-adrenergic antagonist | Treatment of high blood pressure; it improves blood flow and reduces the strain on the heart | Anti-seizure effect | Anti-inflammatory; reduces microglial activation | Animal models: [102] |
| Ibuprofen | Non-steroidal anti-inflammatory drug | Inhibitor of cyclooxygenase 2 | Anti-seizure effect | Reduces activity of post-synaptic NMDA glutamate receptors | Animal models: [103,104,105] |
| N-acetyl Cysteine | Antioxidant | Precursor of reduced glutathione | Anti-seizure effect | Antioxidant, scavenger of reactive oxygen species | Animal models: [84] Humans: [106] |
5.5. Ibuprofen
5.6. N-Acetyl-Cysteine
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xanthos, D.N.; Sandkuhler, J. Neurogenic neuroinflammation: Inflammatory cns reactions in response to neuronal activity. Nat. Rev. Neurosci. 2014, 15, 43–53. [Google Scholar] [CrossRef]
- Dey, A.; Kang, X.; Qiu, J.; Du, Y.; Jiang, J. Anti-inflammatory small molecules to treat seizures and epilepsy: From bench to bedside. Trends Pharmacol. Sci. 2016, 37, 463–484. [Google Scholar] [CrossRef]
- Vezzani, A. Brain inflammation and seizures: Evolving concepts and new findings in the last 2 decades. Epilepsy Curr. 2020, 20, 40S–43S. [Google Scholar] [CrossRef]
- Okin, D.; Medzhitov, R. Evolution of inflammatory diseases. Curr. Biol. 2012, 22, R733–R740. [Google Scholar] [CrossRef]
- Kolliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An integrating overview of reactive-neuroimmune cell interactions in health and disease. Mediators Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef]
- De Nardo, D. Activation of the innate immune receptors: Guardians of the micro galaxy: Activation and functions of the innate immune receptors. Adv. Exp. Med. Biol. 2017, 1024, 1–35. [Google Scholar]
- Klegeris, A. Regulation of neuroimmune processes by damage- and resolution-associated molecular patterns. Neural Regen. Res. 2021, 16, 423–429. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. Damp-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Sanz, P.; Garcia-Gimeno, M.A. Reactive glia inflammatory signaling pathways and epilepsy. Int. J. Mol. Sci. 2020, 21, 4096. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Dokalis, N.; Prinz, M. Resolution of neuroinflammation: Mechanisms and potential therapeutic option. Semin. Immunopathol. 2019, 41, 699–709. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef]
- Augusto-Oliveira, M.; Arrifano, G.P.; Takeda, P.Y.; Lopes-Araujo, A.; Santos-Sacramento, L.; Anthony, D.C.; Verkhratsky, A.; Crespo-Lopez, M.E. Astroglia-specific contributions to the regulation of synapses, cognition and behaviour. Neurosci. Biobehav. Rev. 2020, 118, 331–357. [Google Scholar] [CrossRef]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Munch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Kinboshi, M.; Ikeda, A.; Ohno, Y. Role of astrocytic inwardly rectifying potassium (kir) 4.1 channels in epileptogenesis. Front. Neurol. 2020, 11, 626658. [Google Scholar] [CrossRef]
- Binder, D.K.; Steinhauser, C. Astrocytes and epilepsy. Neurochem. Res. 2021, 46, 2687–2695. [Google Scholar] [CrossRef]
- Vezzani, A.; Ravizza, T.; Bedner, P.; Aronica, E.; Steinhauser, C.; Boison, D. Astrocytes in the initiation and progression of epilepsy. Nat. Rev. Neurol. 2022, 18, 707–722. [Google Scholar] [CrossRef]
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzeriat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef]
- Dejakaisaya, H.; Kwan, P.; Jones, N.C. Astrocyte and glutamate involvement in the pathogenesis of epilepsy in alzheimer’s disease. Epilepsia 2021, 62, 1485–1493. [Google Scholar] [CrossRef]
- Eid, T.; Lee, T.W.; Patrylo, P.; Zaveri, H.P. Astrocytes and glutamine synthetase in epileptogenesis. J. Neurosci. Res. 2019, 97, 1345–1362. [Google Scholar] [CrossRef]
- Sandhu, M.R.S.; Gruenbaum, B.F.; Gruenbaum, S.E.; Dhaher, R.; Deshpande, K.; Funaro, M.C.; Lee, T.W.; Zaveri, H.P.; Eid, T. Astroglial glutamine synthetase and the pathogenesis of mesial temporal lobe epilepsy. Front. Neurol. 2021, 12, 665334. [Google Scholar] [CrossRef]
- Mukhtar, I. Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure 2020, 82, 65–79. [Google Scholar]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Aronica, E.; Ravizza, T.; Zurolo, E.; Vezzani, A. Astrocyte immune responses in epilepsy. Glia 2012, 60, 1258–1268. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Dossi, E.; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef]
- Baulac, M.; de Boer, H.; Elger, C.; Glynn, M.; Kalviainen, R.; Little, A.; Mifsud, J.; Perucca, E.; Pitkanen, A.; Ryvlin, P. Epilepsy priorities in europe: A report of the ilae-ibe epilepsy advocacy europe task force. Epilepsia 2015, 56, 1687–1695. [Google Scholar] [CrossRef]
- Robel, S.; Sontheimer, H. Glia as drivers of abnormal neuronal activity. Nat. Neurosci. 2016, 19, 28–33. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Sun, D.; Tanaka, J. Snapshot of microglial physiological functions. Neurochem. Int. 2021, 144, 104960. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune cell regulation of glia during cns injury and disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef]
- Kinoshita, S.; Koyama, R. Pro- and anti-epileptic roles of microglia. Neural Regen. Res. 2021, 16, 1369–1371. [Google Scholar]
- Robel, S.; Buckingham, S.C.; Boni, J.L.; Campbell, S.L.; Danbolt, N.C.; Riedemann, T.; Sutor, B.; Sontheimer, H. Reactive astrogliosis causes the development of spontaneous seizures. J. Neurosci. 2015, 35, 3330–3345. [Google Scholar] [CrossRef]
- Zhang, D.; Ren, J.; Luo, Y.; He, Q.; Zhao, R.; Chang, J.; Yang, Y.; Guo, Z.N. T cell response in ischemic stroke: From mechanisms to translational insights. Front. Immunol. 2021, 12, 707972. [Google Scholar] [CrossRef]
- Hiragi, T.; Ikegaya, Y.; Koyama, R. Microglia after seizures and in epilepsy. Cells 2018, 7, 26. [Google Scholar] [CrossRef]
- Bernaus, A.; Blanco, S.; Sevilla, A. Glia crosstalk in neuroinflammatory diseases. Front. Cell Neurosci. 2020, 14, 209. [Google Scholar] [CrossRef]
- Borst, K.; Schwabenland, M.; Prinz, M. Microglia metabolism in health and disease. Neurochem. Int. 2019, 130, 104331. [Google Scholar] [CrossRef]
- Lee, J.W.; Chun, W.; Lee, H.J.; Kim, S.M.; Min, J.H.; Kim, D.Y.; Kim, M.O.; Ryu, H.W.; Lee, S.U. The role of microglia in the development of neurodegenerative diseases. Biomedicines 2021, 9, 1449. [Google Scholar] [CrossRef]
- Wong, M.; Guo, D. Dendritic spine pathology in epilepsy: Cause or consequence? Neuroscience 2013, 251, 141–150. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Wyatt-Johnson, S.K.; Brewster, A.L. Emerging roles for microglial phagocytic signaling in epilepsy. Epilepsy Curr. 2020, 20, 33–38. [Google Scholar] [CrossRef]
- Shen, W.; Pristov, J.B.; Nobili, P.; Nikolic, L. Can glial cells save neurons in epilepsy? Neural Regen. Res. 2023, 18, 1417–1422. [Google Scholar]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-astrocyte crosstalk: An intimate molecular conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of microglia and astrocytes in the neurovascular unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte crosstalk in cns inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Eyo, U.B.; Murugan, M.; Wu, L.J. Microglia-neuron communication in epilepsy. Glia 2017, 65, 5–18. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the central nervous system. Cell Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Morin-Brureau, M.; Milior, G.; Royer, J.; Chali, F.; LeDuigou, C.; Savary, E.; Blugeon, C.; Jourdren, L.; Akbar, D.; Dupont, S.; et al. Microglial phenotypes in the human epileptic temporal lobe. Brain 2018, 141, 3343–3360. [Google Scholar] [CrossRef]
- Koh, S. Role of neuroinflammation in evolution of childhood epilepsy. J. Child Neurol. 2018, 33, 64–72. [Google Scholar] [CrossRef]
- Sano, F.; Shigetomi, E.; Shinozaki, Y.; Tsuzukiyama, H.; Saito, K.; Mikoshiba, K.; Horiuchi, H.; Cheung, D.L.; Nabekura, J.; Sugita, K.; et al. Reactive astrocyte-driven epileptogenesis is induced by microglia initially activated following status epilepticus. JCI Insight 2021, 6, e135391. [Google Scholar] [CrossRef]
- Passaro, A.P.; Lebos, A.L.; Yao, Y.; Stice, S.L. Immune response in neurological pathology: Emerging role of central and peripheral immune crosstalk. Front. Immunol. 2021, 12, 676621. [Google Scholar] [CrossRef]
- Cervellati, C.; Trentini, A.; Pecorelli, A.; Valacchi, G. Inflammation in neurological disorders: The thin boundary between brain and periphery. Antioxid. Redox Signal 2020, 33, 191–210. [Google Scholar] [CrossRef]
- Yamanaka, G.; Morichi, S.; Takamatsu, T.; Watanabe, Y.; Suzuki, S.; Ishida, Y.; Oana, S.; Yamazaki, T.; Takata, F.; Kawashima, H. Links between immune cells from the periphery and the brain in the pathogenesis of epilepsy: A narrative review. Int. J. Mol. Sci. 2021, 22, 4395. [Google Scholar] [CrossRef]
- Qiu, Y.M.; Zhang, C.L.; Chen, A.Q.; Wang, H.L.; Zhou, Y.F.; Li, Y.N.; Hu, B. Immune cells in the bbb disruption after acute ischemic stroke: Targets for immune therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef]
- Rodriguez Murua, S.; Farez, M.F.; Quintana, F.J. The immune response in multiple sclerosis. Annu. Rev. Pathol. 2022, 17, 121–139. [Google Scholar] [CrossRef]
- Pitsch, J.; van Loo, K.M.J.; Gallus, M.; Dik, A.; Kamalizade, D.; Baumgart, A.K.; Gnatkovsky, V.; Muller, J.A.; Opitz, T.; Hicking, G.; et al. Cd8(+) t-lymphocyte-driven limbic encephalitis results in temporal lobe epilepsy. Ann. Neurol. 2021, 89, 666–685. [Google Scholar] [CrossRef]
- Troscher, A.R.; Sakaraki, E.; Mair, K.M.; Kock, U.; Racz, A.; Borger, V.; Cloppenborg, T.; Becker, A.J.; Bien, C.G.; Bauer, J. T cell numbers correlate with neuronal loss rather than with seizure activity in medial temporal lobe epilepsy. Epilepsia 2021, 62, 1343–1353. [Google Scholar] [CrossRef]
- Villasana-Salazar, B.; Vezzani, A. Neuroinflammation microenvironment sharpens seizure circuit. Neurobiol. Dis. 2023, 178, 106027. [Google Scholar] [CrossRef]
- Vezzani, A.; Di Sapia, R.; Kebede, V.; Balosso, S.; Ravizza, T. Neuroimmunology of status epilepticus. Epilepsy Behav. 2023, 140, 109095. [Google Scholar] [CrossRef]
- Chen, Y.; Nagib, M.M.; Yasmen, N.; Sluter, M.N.; Littlejohn, T.L.; Yu, Y.; Jiang, J. Neuroinflammatory mediators in acquired epilepsy: An update. Inflamm. Res. 2023, 72, 683–701. [Google Scholar] [CrossRef]
- Kaminska, B.; Mota, M.; Pizzi, M. Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim. Biophys. Acta 2016, 1862, 339–351. [Google Scholar] [CrossRef]
- Lee, T.S.; Li, A.Y.; Rapuano, A.; Mantis, J.; Eid, T.; Seyfried, T.N.; de Lanerolle, N.C. Gene expression in the epileptic (el) mouse hippocampus. Neurobiol. Dis. 2020, 147, 105152. [Google Scholar] [CrossRef]
- Grote, A.; Heiland, D.H.; Taube, J.; Helmstaedter, C.; Ravi, V.M.; Will, P.; Hattingen, E.; Schure, J.R.; Witt, J.A.; Reimers, A.; et al. ‘Hippocampal innate inflammatory gliosis only’ in pharmacoresistant temporal lobe epilepsy. Brain 2023, 146, 549–560. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and molecular mechanisms of metformin action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Sanz, P.; Serratosa, J.M.; Sanchez, M.P. Beneficial effects of metformin on the central nervous system, with a focus on epilepsy and lafora disease. Int. J. Mol. Sci. 2021, 22, 5351. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the insulin sensitizer metformin in alzheimer disease: Pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Brakedal, B.; Haugarvoll, K.; Tzoulis, C. Simvastatin is associated with decreased risk of parkinson disease. Ann. Neurol. 2017, 81, 329–330. [Google Scholar] [CrossRef]
- Hervas, D.; Fornes-Ferrer, V.; Gomez-Escribano, A.P.; Sequedo, M.D.; Peiro, C.; Millan, J.M.; Vazquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef]
- Abdi, M.; Pasbakhsh, P.; Shabani, M.; Nekoonam, S.; Sadeghi, A.; Fathi, F.; Abouzaripour, M.; Mohamed, W.; Zibara, K.; Kashani, I.R.; et al. Metformin therapy attenuates pro-inflammatory microglia by inhibiting nf-kappab in cuprizone demyelinating mouse model of multiple sclerosis. Neurotox. Res. 2021, 39, 1732–1746. [Google Scholar] [CrossRef]
- Wang, Y.W.; He, S.J.; Feng, X.; Cheng, J.; Luo, Y.T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Dev. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Nandini, H.S.; Paudel, Y.N.; Krishna, K.L. Envisioning the neuroprotective effect of metformin in experimental epilepsy: A portrait of molecular crosstalk. Life Sci. 2019, 233, 116686. [Google Scholar]
- Yimer, E.M.; Surur, A.; Wondafrash, D.Z.; Gebre, A.K. The effect of metformin in experimentally induced animal models of epileptic seizure. Behav. Neurol. 2019, 2019, 6234758. [Google Scholar] [CrossRef]
- Berthier, A.; Paya, M.; Garcia-Cabrero, A.M.; Ballester, M.I.; Heredia, M.; Serratosa, J.M.; Sanchez, M.P.; Sanz, P. Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of lafora disease. Mol. Neurobiol. 2016, 53, 1296–1309. [Google Scholar] [CrossRef]
- Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanz, P.; Sanchez, M.P. 4-phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol-induced seizures in a malin knockout model of lafora disease. Neuroreport 2017, 28, 268–271. [Google Scholar] [CrossRef]
- Burgos, D.F.; Machio-Castello, M.; Iglesias-Cabeza, N.; Giraldez, B.G.; Gonzalez-Fernandez, J.; Sanchez-Martin, G.; Sanchez, M.P.; Serratosa, J.M. Early treatment with metformin improves neurological outcomes in lafora disease. Neurotherapeutics 2023, 20, 230–244. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Y.; Li, F.; Ling, L. Sphingosine-1-phosphate receptor modulators in stroke treatment. J. Neurochem. 2022, 162, 390–403. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Willis, C.M.; Hamel, R.; Krzak, G.; Pluchino, S. Metabolic control of smoldering neuroinflammation. Front. Immunol. 2021, 12, 705920. [Google Scholar] [CrossRef]
- Hajipour, S.; Khombi Shooshtari, M.; Farbood, Y.; Ali Mard, S.; Sarkaki, A.; Moradi Chameh, H.; Sistani Karampour, N.; Ghafouri, S. Fingolimod administration following hypoxia induced neonatal seizure can restore impaired long-term potentiation and memory performance in adult rats. Neuroscience 2023, 519, 107–119. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Gnatkovsky, V.; Othman, I.; Shaikh, M.F. From the molecular mechanism to pre-clinical results: Anti-epileptic effects of fingolimod. Curr. Neuropharmacol. 2020, 18, 1126–1137. [Google Scholar] [CrossRef]
- Klein, P.; Friedman, A.; Hameed, M.Q.; Kaminski, R.M.; Bar-Klein, G.; Klitgaard, H.; Koepp, M.; Jozwiak, S.; Prince, D.A.; Rotenberg, A.; et al. Repurposed molecules for antiepileptogenesis: Missing an opportunity to prevent epilepsy? Epilepsia 2020, 61, 359–386. [Google Scholar] [CrossRef]
- Pournajaf, S.; Dargahi, L.; Javan, M.; Pourgholami, M.H. Molecular pharmacology and novel potential therapeutic applications of fingolimod. Front. Pharmacol. 2022, 13, 807639. [Google Scholar] [CrossRef]
- Yang, L.X.; Yao, Y.Y.; Yang, J.R.; Cheng, H.L.; Zhu, X.J.; Zhang, Z.J. Sphingosine 1-phosphate receptor 1 regulates blood-brain barrier permeability in epileptic mice. Neural Regen. Res. 2023, 18, 1763–1769. [Google Scholar] [CrossRef]
- Rubio, T.; Campos-Rodriguez, A.; Sanz, P. Beneficial effect of fingolimod in a lafora disease mouse model by preventing reactive astrogliosis-derived neuroinflammation and brain infiltration of t-lymphocytes. Mol. Neurobiol. 2023. [CrossRef]
- Naegelin, Y.; Kuhle, J.; Schadelin, S.; Datta, A.N.; Magon, S.; Amann, M.; Barro, C.; Ramelli, G.P.; Heesom, K.; Barde, Y.A.; et al. Fingolimod in children with rett syndrome: The fingorett study. Orphanet J. Rare Dis. 2021, 16, 19. [Google Scholar] [CrossRef]
- Rosito, M.; Testi, C.; Parisi, G.; Cortese, B.; Baiocco, P.; Di Angelantonio, S. Exploring the use of dimethyl fumarate as microglia modulator for neurodegenerative diseases treatment. Antioxidants 2020, 9, 700. [Google Scholar] [CrossRef]
- Sadovnikova, I.S.; Gureev, A.P.; Ignatyeva, D.A.; Gryaznova, M.V.; Chernyshova, E.V.; Krutskikh, E.P.; Novikova, A.G.; Popov, V.N. Nrf2/are activators improve memory in aged mice via maintaining of mitochondrial quality control of brain and the modulation of gut microbiome. Pharmaceuticals 2021, 14, 607. [Google Scholar] [CrossRef]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 2006, 145, 101–107. [Google Scholar] [CrossRef]
- Lim, J.L.; van der Pol, S.M.; Di Dio, F.; van Het Hof, B.; Kooij, G.; de Vries, H.E.; van Horssen, J. Protective effects of monomethyl fumarate at the inflamed blood-brain barrier. Microvasc. Res. 2016, 105, 61–69. [Google Scholar] [CrossRef]
- Vainio, S.K.; Dickens, A.M.; Matilainen, M.; Lopez-Picon, F.R.; Aarnio, R.; Eskola, O.; Solin, O.; Anthony, D.C.; Rinne, J.O.; Airas, L.; et al. Dimethyl fumarate decreases short-term but not long-term inflammation in a focal eae model of neuroinflammation. EJNMMI Res. 2022, 12, 6. [Google Scholar] [CrossRef]
- Singh, N.; Vijayanti, S.; Saha, L.; Bhatia, A.; Banerjee, D.; Chakrabarti, A. Neuroprotective effect of nrf2 activator dimethyl fumarate, on the hippocampal neurons in chemical kindling model in rat. Epilepsy Res. 2018, 143, 98–104. [Google Scholar] [CrossRef]
- Singh, N.; Saha, L.; Kumari, P.; Singh, J.; Bhatia, A.; Banerjee, D.; Chakrabarti, A. Effect of dimethyl fumarate on neuroinflammation and apoptosis in pentylenetetrazol kindling model in rats. Brain Res. Bull. 2019, 144, 233–245. [Google Scholar] [CrossRef]
- Sandouka, S.; Singh, P.K.; Saadi, A.; Taiwo, R.O.; Sheeni, Y.; Zhang, T.; Deeb, L.; Guignet, M.; White, S.H.; Shekh-Ahmad, T. Repurposing dimethyl fumarate as an antiepileptogenic and disease-modifying treatment for drug-resistant epilepsy. J. Transl. Med. 2023, 21, 796. [Google Scholar] [CrossRef]
- Delpech, J.C.; Madore, C.; Nadjar, A.; Joffre, C.; Wohleb, E.S.; Laye, S. Microglia in neuronal plasticity: Influence of stress. Neuropharmacology 2015, 96, 19–28. [Google Scholar] [CrossRef]
- Kota, D.J.; Prabhakara, K.S.; van Brummen, A.J.; Bedi, S.; Xue, H.; DiCarlo, B.; Cox, C.S., Jr.; Olson, S.D. Propranolol and mesenchymal stromal cells combine to treat traumatic brain injury. Stem Cells Transl. Med. 2016, 5, 33–44. [Google Scholar] [CrossRef]
- Lin, T.W.; Harward, S.C.; Huang, Y.Z.; McNamara, J.O. Targeting bdnf/trkb pathways for preventing or suppressing epilepsy. Neuropharmacology 2020, 167, 107734. [Google Scholar] [CrossRef]
- Woiciechowsky, C.; Schoning, B.; Stoltenburg-Didinger, G.; Stockhammer, F.; Volk, H.D. Brain-il-1 beta triggers astrogliosis through induction of il-6: Inhibition by propranolol and il-10. Med. Sci. Monit. 2004, 10, BR325–BR330. [Google Scholar]
- Dobarro, M.; Gerenu, G.; Ramirez, M.J. Propranolol reduces cognitive deficits, amyloid and tau pathology in alzheimer’s transgenic mice. Int. J. Neuropsychopharmacol. 2013, 16, 2245–2257. [Google Scholar] [CrossRef]
- Molla, B.; Heredia, M.; Sanz, P. Modulators of neuroinflammation have a beneficial effect in a lafora disease mouse model. Mol. Neurobiol. 2021, 58, 2508–2522. [Google Scholar] [CrossRef]
- Liu, R.; Wu, S.; Guo, C.; Hu, Z.; Peng, J.; Guo, K.; Zhang, X.; Li, J. Ibuprofen exerts antiepileptic and neuroprotective effects in the rat model of pentylenetetrazol-induced epilepsy via the cox-2/nlrp3/il-18 pathway. Neurochem. Res. 2020, 45, 2516–2526. [Google Scholar] [CrossRef]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alnaaim, S.A.; Alexiou, A.; Papadakis, M.; Saad, H.M.; Batiha, G.E. Autophagy and autophagy signaling in epilepsy: Possible role of autophagy activator. Mol. Med. 2023, 29, 142. [Google Scholar] [CrossRef]
- Sinha, P.; Verma, B.; Ganesh, S. Age-dependent reduction in the expression levels of genes involved in progressive myoclonus epilepsy correlates with increased neuroinflammation and seizure susceptibility in mouse models. Mol. Neurobiol. 2022, 59, 5532–5548. [Google Scholar] [CrossRef]
- Hurd, R.W.; Wilder, B.J.; Helveston, W.R.; Uthman, B.M. Treatment of four siblings with progressive myoclonus epilepsy of the unverricht-lundborg type with n-acetylcysteine. Neurology 1996, 47, 1264–1268. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz, P.; Rubio, T.; Garcia-Gimeno, M.A. Neuroinflammation and Epilepsy: From Pathophysiology to Therapies Based on Repurposing Drugs. Int. J. Mol. Sci. 2024, 25, 4161. https://doi.org/10.3390/ijms25084161
Sanz P, Rubio T, Garcia-Gimeno MA. Neuroinflammation and Epilepsy: From Pathophysiology to Therapies Based on Repurposing Drugs. International Journal of Molecular Sciences. 2024; 25(8):4161. https://doi.org/10.3390/ijms25084161
Chicago/Turabian StyleSanz, Pascual, Teresa Rubio, and Maria Adelaida Garcia-Gimeno. 2024. "Neuroinflammation and Epilepsy: From Pathophysiology to Therapies Based on Repurposing Drugs" International Journal of Molecular Sciences 25, no. 8: 4161. https://doi.org/10.3390/ijms25084161
APA StyleSanz, P., Rubio, T., & Garcia-Gimeno, M. A. (2024). Neuroinflammation and Epilepsy: From Pathophysiology to Therapies Based on Repurposing Drugs. International Journal of Molecular Sciences, 25(8), 4161. https://doi.org/10.3390/ijms25084161