
Another Use for a Proven Drug: Experimental Evidence for the Potential of Artemisinin and Its Derivatives to Treat Alzheimer’s Disease


Abstract
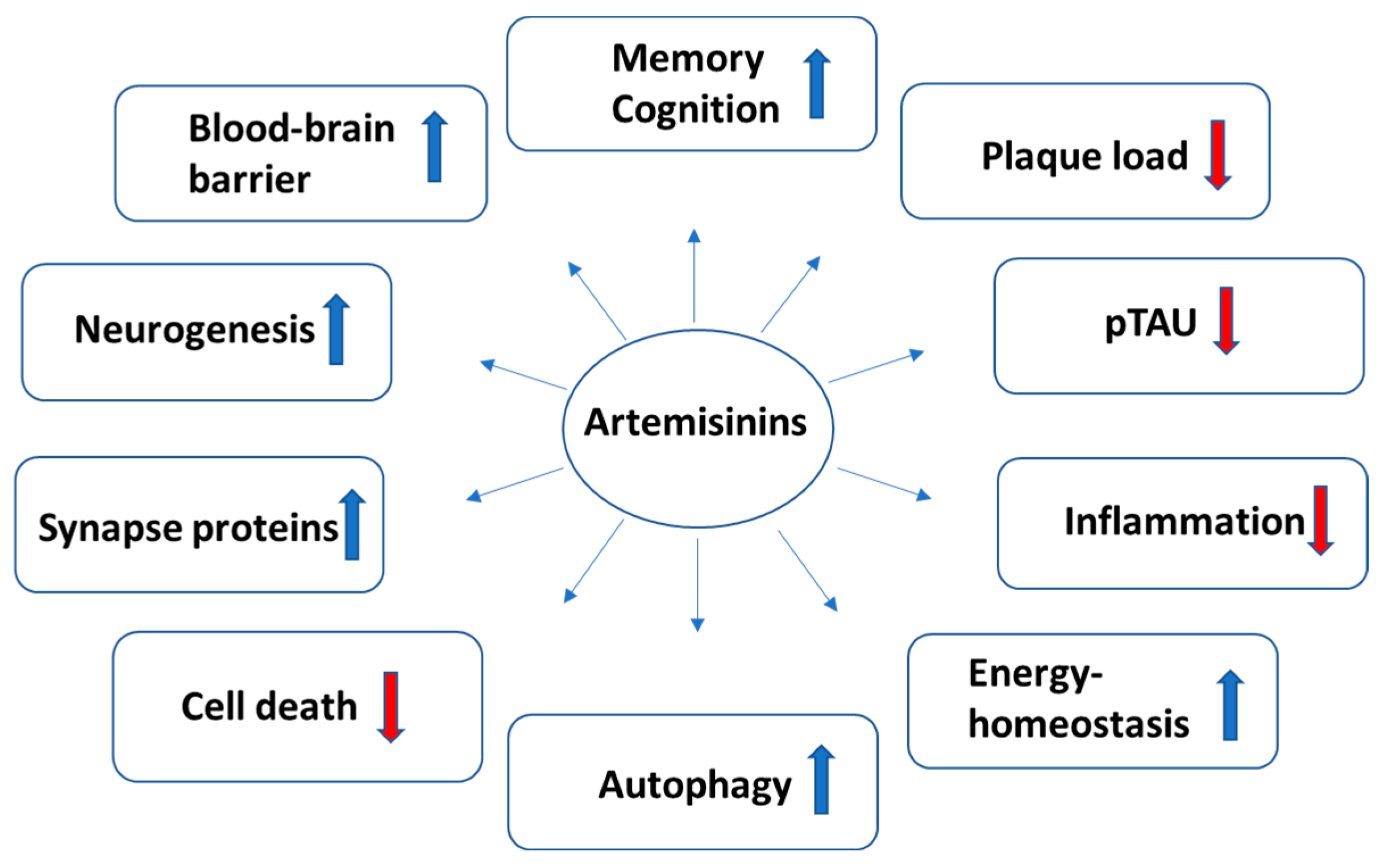
:1. Introduction
2. Artemisinins: Chemical Structure, Pharmacokinetic Behaviors and Antimalaria Activities
3. Alzheimer’s Disease Mouse Models Used for Testing Artemisinins
4. Effects of Artemisinins on Hallmarks of AD Pathogenesis
4.1. Aβ Pathology—Plaque Formation
4.2. Tau Pathology
4.3. Inflammation
4.4. Oxidative Stress and Mitochondrial Dysfunction
4.5. Autophagy
4.6. Cell Death
4.7. Synapse Pathology
4.8. Neurogenesis
4.9. Blood–Brain Barrier
4.10. Memory and Cognition
5. Artemisinins Toxicity
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W.; Styren, S.D. Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration 1996, 5, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Mecca, A.P.; O’Dell, R.S.; Sharp, E.S.; Banks, E.R.; Bartlett, H.H.; Zhao, W.; Lipior, S.; Diepenbrock, N.G.; Chen, M.K.; Naganawa, M.; et al. Synaptic density and cognitive performance in Alzheimer’s disease: A PET imaging study with [11C]UCB-J. Alzheimers Dement. 2022, 18, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J. 2017, 284, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The amyloid cascade hypothesis: An updated critical review. Brain 2023, 146, 3969–3990. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Sig. Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What Happens with the Circuit in Alzheimer’s Disease in Mice and Humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.A.; Suh, Y.H. Pathophysiological roles of amyloidogenic carboxy-terminal fragments of the beta-amyloid precursor protein in Alzheimer’s disease. J. Pharmacol. Sci. 2005, 97, 461–471. [Google Scholar] [CrossRef]
- Xu, Y.; Kim, H.S.; Joo, Y.; Choi, Y.; Chang, K.A.; Park, C.H.; Shin, K.Y.; Kim, S.; Cheon, Y.H.; Baik, T.K.; et al. Intracellular domains of amyloid precursor-like protein 2 interact with CP2 transcription factor in the nucleus and induce glycogen synthase kinase-3beta expression. Cell Death Differ. 2007, 14, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef] [PubMed]
- van der Kant, R.; Goldstein, L.S. Cellular functions of the amyloid precursor protein from development to dementia. Dev. Cell 2015, 32, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Y.; Mattson, M.P. Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp. Neurol. 1994, 128, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.C.; Ludewig, S.; Winschel, A.; Abel, T.; Bold, C.; Salzburger, L.R.; Klein, S.; Han, K.; Weyer, S.W.; Fritz, A.K.; et al. Distinct in vivo roles of secreted APP ectodomain variants APPsα and APPsβ in regulation of spine density, synaptic plasticity, and cognition. EMBO J. 2018, 37, e98335. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Palmeri, A. Hormetic effect of amyloid-β peptide in synaptic plasticity and memory. Neurobiol. Aging 2012, 33, 1484.e15–1484.e24. [Google Scholar] [CrossRef]
- Coronel, R.; Palmer, C.; Bernabeu-Zornoza, A.; Monteagudo, M.; Rosca, A.; Zambrano, A.; Liste, I. Physiological effects of amyloid precursor protein and its derivatives on neural stem cell biology and signaling pathways involved. Neural Regen. Res. 2019, 14, 1661–1671. [Google Scholar]
- Zimbone, S.; Monaco, I.; Gianì, F.; Pandini, G.; Copani, A.G.; Giuffrida, M.L.; Rizzarelli, E. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 2018, 17, e12684. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Bishop, G.M.; Perry, G.; Smith, M.A. Amyloid-beta: A vascular sealant that protects against hemorrhage? J. Neurosci. Res. 2002, 70, 356. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.S.; Li, S.; Zhang, F.; Deng, J.; Zeng, L.H.; Tan, J. Amyloid Precursor Protein: A Regulatory Hub in Alzheimer’s Disease. Aging Dis. 2023, 15, 201–225. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.C.; Lin, G.A.; Whittington, M.D.; Agboola, F.; Herron-Smith, S.; Rind, D.; Pearson, S.D. The effectiveness and value of lecanemab for early Alzheimer disease: A summary from the Institute for Clinical and Economic Review’s California Technology Assessment Forum. J. Manag. Care Spec. Pharm. 2023, 29, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Rahman, M.S.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M. Revisiting the Amyloid Cascade Hypothesis: From Anti-Aβ Therapeutics to Auspicious New Ways for Alzheimer’s. Disease. Int. J. Mol. Sci. 2020, 21, 5858. [Google Scholar] [CrossRef] [PubMed]
- Rivas-García, L.; Quiles, J.L.; Roma-Rodrigues, C.; Raposo, L.R.; Navarro-Hortal, M.D.; Romero-Márquez, J.M.; Esteban-Muñoz, A.; Varela-López, A.; García, L.C.; Cianciosi, D.; et al. Rosa x hybrida extracts with dual actions: Antiproliferative effects against tumour cells and inhibitor of Alzheimer disease. Food Chem. Toxicol. 2021, 149, 112018. [Google Scholar] [CrossRef]
- Rivas-García, L.; Romero-Márquez, J.M.; Navarro-Hortal, M.D.; Esteban-Muñoz, A.; Giampieri, F.; Sumalla-Cano, S.; Battino, M.; Quiles, J.L.; Llopis, J.; Sánchez-González, C. Unravelling potential biomedical applications of the edible flower Tulbaghia violacea. Food Chem. 2022, 381, 132096. [Google Scholar] [CrossRef]
- Romero-Márquez, J.M.; Forbes-Hernández, T.Y.; Navarro-Hortal, M.D.; Quirantes-Piné, R.; Grosso, G.; Giampieri, F.; Lipari, V.; Sánchez-González, C.; Battino, M.; Quiles, J.L. Molecular Mechanisms of the Protective Effects of Olive Leaf Polyphenols against Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 4353. [Google Scholar] [CrossRef]
- Navarro-Hortal, M.D.; Romero-Márquez, J.M.; Muñoz-Ollero, P.; Jiménez-Trigo, V.; Esteban-Muñoz, A.; Tutusaus, K.; Giampieri, F.; Battino, M.; Sánchez-González, C.; Rivas-García, L.; et al. Amyloid β-but not Tau-induced neurotoxicity is suppressed by Manuka honey via HSP-16.2 and SKN-1/Nrf2 pathways in an in vivo model of Alzheimer’s disease. Food Funct. 2022, 13, 11185–11199. [Google Scholar] [CrossRef]
- Romero-Márquez, J.M.; Navarro-Hortal, M.D.; Orantes, F.J.; Esteban-Muñoz, A.; Pérez-Oleaga, C.M.; Battino, M.; Sánchez-González, C.; Rivas-García, L.; Giampieri, F.; Quiles, J.L.; et al. In Vivo Anti-Alzheimer and Antioxidant Properties of Avocado (Persea americana Mill.) Honey from Southern Spain. Antioxidants 2023, 12, 404. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Hasana, S.; Ahmad, J.; Hossain, M.F.; Rahman, M.M.; Behl, T.; Rauf, A.; Ahmad, A.; Hafeez, A.; Perveen, A.; et al. Anti-Neuroinflammatory Potential of Polyphenols by Inhibiting NF-κB to Halt Alzheimer’s Disease. Curr. Pharm. Des. 2021, 27, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014, 142, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.J.; Fischer, N.; Efferth, T. Phytochemicals as inhibitors of NF-κB for treatment of Alzheimer’s disease. Pharmacol. Res. 2018, 129, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Rubin, L.; Silva, M.; Li, S.; Yang, C.; Lazarovici, P.; Zheng, W. Current Progress on Neuroprotection Induced by Artemisia, Ginseng, Astragalus, and Ginkgo Traditional Chinese Medicines for the Therapy of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2022, 14, 3777021. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Sun, Y.; Su, R.; Gao, H. Anti-Alzheimer’s disease potential of traditional chinese medicinal herbs as inhibitors of BACE1 and AChE enzymes. Biomed Pharmacother. 2022, 154, 113576. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Bustamante, L.; Haynes, R.K.; Staines, H.M. Artemisinins: Their growing importance in medicine. Trends Pharmacol. Sci. 2008, 29, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Li, H.; Yang, Y.; Hou, L. Anti-inflammatory and immunoregulatory functions of artemisinin and its derivatives. Mediat. Inflamm. 2015, 2015, 435713. [Google Scholar] [CrossRef] [PubMed]
- Navaratnam, V.; Mansor, S.M.; Sit, N.W.; Grace, J.; Li, Q.; Olliaro, P. Pharmacokinetics of artemisinin-type compounds. Clin. Pharmacokinet. 2000, 39, 255–270. [Google Scholar] [CrossRef]
- Fu, C.; Shi, H.; Chen, H.; Zhang, K.; Wang, M.; Qiu, F. Oral Bioavailability Comparison of Artemisinin, Deoxyartemisinin, and 10-Deoxoartemisinin Based on Computer Simulations and Pharmacokinetics in Rats. ACS Omega 2020, 6, 889–899. [Google Scholar] [CrossRef]
- de Vries, P.J.; Dien, T.K. Clinical pharmacology and therapeutic potential of artemisinin and its derivatives in the treatment of malaria. Drugs 1996, 52, 818–836. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.S.; Fleckenstein, L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011, 10, 263. [Google Scholar] [CrossRef]
- Gautam, A.; Ahmed, T.; Batra, V.; Paliwal, J. Pharmacokinetics and pharmacodyamics of endoperoxide antimalarials. Curr. Drug Metab. 2009, 10, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Aweeka, F.T.; German, P.I. Clinical Pharmacology of Artemisinin-Based Combination Therapies. Clin. Pharmacokinet. 2008, 47, 91–102. [Google Scholar] [CrossRef]
- Lu, F.; He, X.-L.; Richard, C.; Cao, J. A brief history of artemisinin: Modes of action and mechanisms of resistance. Chin. J. Nat. Med. 2019, 17, 331–336. [Google Scholar] [CrossRef]
- Ribbiso, K.A.; Heller, L.E.; Taye, A.; Julian, E.; Willems, A.V.; Roepe, P.D. Artemisinin-Based Drugs Target the Plasmodium falciparum Heme Detoxification Pathway. Antimicrob. Agents Chemother. 2021, 65, e02137-20. [Google Scholar] [CrossRef]
- Bridgford, J.L.; Xie, S.C.; Cobbold, S.A.; Pasaje, C.F.A.; Herrmann, S.; Yang, T.; Gillett, D.L.; Dick, L.R.; Ralph, S.A.; Dogovski, C.; et al. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat. Commun. 2018, 9, 3801. [Google Scholar] [CrossRef] [PubMed]
- Posadino, A.M.; Giordo, R.; Pintus, G.; Mohammed, S.A.; Orhan, I.E.; Fokou, P.V.T.; Sharopov, F.; Adetunji, C.O.; Gulsunoglu-Konuskan, Z.; Ydyrys, A.; et al. Medicinal and mechanistic overview of artemisinin in the treatment of human diseases. Biomed. Pharmacother. 2023, 163, 114866. [Google Scholar] [CrossRef]
- Yuan, D.S.; Chen, Y.P.; Tan, L.L.; Huang, S.Q.; Li, C.Q.; Wang, Q.; Zeng, Q.P. Artemisinin: A Panacea Eligible for Unrestrictive Use? Front. Pharmacol. 2017, 8, 737. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.J.; Vesterinen, H.M.; Beglopoulos, V.; Sena, E.S.; Macleod, M.R. From a mouse: Systematic analysis reveals limitations of experiments testing interventions in Alzheimer’s disease mouse models. Evid.-Based Preclin. Med. 2016, 3, e00015. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J. Alzheimer’s Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Söllvander, S.; Nikitidou, E.; Gallasch, L.; Zyśk, M.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J. Neuroinflamm. 2018, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Rasmussen, J.; Kaeser, S.A.; Marzesco, A.M.; Obermüller, U.; Mahler, J.; Schelle, J.; Odenthal, J.; Krüger, C.; Fritschi, S.K.; et al. Aβ seeding potency peaks in the early stages of cerebral β-amyloidosis. EMBO Rep. 2017, 18, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Slunt, H.H.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol. Aging 2004, 25, 885–892. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Robbins, E.M.; Zhang-Nunes, S.X.; Purcell, S.M.; Betensky, R.A.; Raju, S.; Prada, C.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 2006, 24, 516–524. [Google Scholar] [CrossRef]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.; Verveer, M.; de Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 2012, 7, e42823. [Google Scholar] [CrossRef]
- Jackson, R.J.; Rudinskiy, N.; Herrmann, A.G.; Croft, S.; Kim, J.M.; Petrova, V.; Ramos-Rodriguez, J.J.; Pitstick, R.; Wegmann, S.; Garcia-Alloza, M.; et al. Human tau increases amyloid β plaque size but not amyloid β-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2016, 44, 3056–3066. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van, E.L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.; Barron, A.M.; Brown, M.A.; Abbiati, F.; Carrero, P.; Pike, C.J.; Garcia-Segura, L.M.; Melcangi, R.C. Age-related changes in neuroactive steroid levels in 3xTg-AD mice. Neurobiol. Aging 2013, 34, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Javonillo, D.I.; Tran, K.M.; Phan, J.; Hingco, E.; Kramár, E.A.; da Cunha, C.; Forner, S.; Kawauchi, S.; Milinkeviciute, G.; Gomez-Arboledas, A.; et al. Systematic Phenotyping and Characterization of the 3xTg-AD Mouse Model of Alzheimer’s Disease. Front. Neurosc. 2022, 15, 785276. [Google Scholar] [CrossRef]
- Kitazawa, M.; Medeiros, R.; Laferla, F.M. Transgenic mouse models of Alzheimer disease: Developing a better model as a tool for therapeutic interventions. Curr. Pharm. Des. 2012, 18, 1131–1147. [Google Scholar] [CrossRef] [PubMed]
- Chambon, C.; Wegener, N.; Gravius, A.; Danysz, W. Behavioural and cellular effects of exogenous amyloid-β peptides in rodents. Behav Brain Res. 2011, 225, 623–641. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Cummings, J.; Osse, A.M.L.; Cammann, D.; Powell, J.; Chen, J. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer’s Disease. BioDrugs 2024, 38, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Zhang, C.C.; Sun, X.L.; Cheng, X.X.; Wang, J.B.; Zhang, Y.D.; Xu, J.; Zou, H.Q. Antimalarial drug artemisinin extenuates amyloidogenesis and neuroinflammation in APPswe/PS1dE9 transgenic mice via inhibition of nuclear factor-κB and NLRP3 inflammasome activation. CNS Neurosci. Ther. 2013, 19, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Long, Z.; Ding, Y.; Jiang, T.; Liu, J.; Li, Y.; Liu, Y.; Peng, X.; Wang, K.; Feng, M.; et al. Dihydroartemisinin Ameliorates Learning and Memory in Alzheimer’s Disease Through Promoting Autophagosome-Lysosome Fusion and Autolysosomal Degradation for Aβ Clearance. Front. Aging Neurosci. 2020, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Xiang, W.; Chen, Y.; Peng, N.; Du, X.; Lu, S.; Zuo, Y.; Li, B.; Hu, Y.; Li, X. DHA Ameliorates Cognitive Ability, Reduces Amyloid Deposition, and Nerve Fiber Production in Alzheimer’s Disease. Front. Nutr. 2022, 9, 852433. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.R.; Ma, C.Q.; Jiang, J.H.; Wang, D.P.; Zhang, Q.Q.; Liu, M.R.; Zhao, H.R.; Fang, Q.; Liu, Y. Artesunate restores mitochondrial fusion-fission dynamics and alleviates neuronal injury in Alzheimer’s disease models. J. Neurochem. 2022, 162, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Sagare, A.P.; Lazic, D.; Bazzi, S.; Lawson, E.; Hsu, C.J.; Wang, Y.; Ramanathan, A.; Nelson, A.R.; Zhao, Z.; et al. Anti-malaria drug artesunate prevents development of amyloid-β pathology in mice by upregulating PICALM at the blood-brain barrier. Mol. Neurodegener. 2023, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Kins, S.; Zöller, Y.; Schilling, S.; Gorgas, K.; Groß, D.; Schlicksupp, A.; Rosner, R.; Kirsch, J.; Kuhse, J. Artesunate restores the levels of inhibitory synapse proteins and reduces amyloid-β and C-terminal fragments (CTFs) of the amyloid precursor protein in an AD-mouse model. Mol. Cell. Neurosci. 2021, 113, 103624. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef]
- Pooler, A.M.; Polydoro, M.; Wegmann, S.; Nicholls, S.B.; Spires-Jones, T.L.; Hyman, B.T. Propagation of tau pathology in Alzheimer’s disease: Identification of novel therapeutic targets. Alzheimer’s Res. Ther. 2013, 5, 49. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, X.; Lazarovici, P.; Zheng, W. Artemether Activation of AMPK/GSK3β(ser9)/Nrf2 Signaling Confers Neuroprotection towards β-Amyloid-Induced Neurotoxicity in 3xTg Alzheimer’s Mouse Model. Oxidative Med. Cell. Longev. 2019, 2019, 1862437. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Wang, T.Z.; Hou, C.; Liu, H.Y.; Zhang, Y.; Xue, Z.Z.; Cai, Q.C.; Chen, D.M.; Gao, C.W.; Yang, J.L.; et al. Artemether Attenuates Aβ25-35-Induced Cognitive Impairments by Downregulating Aβ, BACE1, mTOR and Tau Proteins. Clin. Lab. 2021, 67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, S.; Gaur, U.; Zheng, W. Artemisinin Improved Neuronal Functions in Alzheimer’s Disease Animal Model 3xtg Mice and Neuronal Cells via Stimulating the ERK/CREB Signaling Pathway. Aging Dis. 2020, 11, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Lei, B.; Yang, C.; Silva, M.; Xing, X.; Yu, H.; Lu, J.; Zheng, W. Artemisia annua Extract Improves the Cognitive Deficits and Reverses the Pathological Changes of Alzheimer’s Disease via Regulating YAP Signaling. Int. J. Mol. Sci. 2023, 24, 5259. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.R.; Touchard, S.; Leckey, C.; O’Hagan, C.; Nevado-Holgado, A.J.; NIMAConsortium Barkhof, F.; Bertram, L.; Blin, O.; Bos, I.; Dobricic, V.; et al. Annex: NIMA–Wellcome Trust Consortium for Neuroimmunology of Mood Disorders and Alzheimer’s Disease. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Carret-Rebillat, A.S.; Pace, C.; Gourmaud, S.; Ravasi, L.; Montagne-Stora, S.; Longueville, S.; Tible, M.; Sudol, E.; Chang, R.C.; Paquet, C.; et al. Neuroinflammation and Aβ accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation. Sci. Rep. 2015, 5, 8489. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Jha, D.; Bakker, E.N.T.P.; Kumar, R. Mechanistic and therapeutic role of NLRP3 inflammasome in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2023. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Cai, W.; Yang, Q.; Yang, L.; Dai, Y.; Zhao, Z.; Yin, J.; Li, Y.; Li, Q.; Wang, Y.; et al. Artemisinin B Improves Learning and Memory Impairment in AD Dementia Mice by Suppressing Neuroinflammation. Neuroscience 2018, 395, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, X.; Yang, C.; Jiang, Y.; Zhou, W.; Zheng, W. Artemisinin Attenuates Amyloid-Induced Brain Inflammation and Memory Impairments by Modulating TLR4/NF-κB Signaling. Int. J. Mol. Sci. 2022, 23, 6354. [Google Scholar] [CrossRef] [PubMed]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Nikolaeva, N.S.; Yandulova, E.Y.; Aleksandrova, Y.R.; Starikov, A.S.; Neganova, M.E. The Role of a Pathological Interaction between β-amyloid and Mitochondria in the Occurrence and Development of Alzheimer’s Disease. Acta Naturae 2022, 14, 19–34. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. Int. J. Mol. Sci. 2023, 24, 3739. [Google Scholar] [CrossRef] [PubMed]
- Poorgholam, P.; Yaghmaei, P.; Noureddini, M.; Hajebrahimi, Z. Effects of artemisinin and TSP-1-human endometrial-derived stem cells on a streptozocin-induced model of Alzheimer’s disease and diabetes in Wistar rats. Acta Neurobiol. Exp. 2021, 81, 141–150. [Google Scholar]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic exposure to sub-lethal beta-amyloid (Abeta) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef]
- Bhatia, S.; Rawal, R.; Sharma, P.; Singh, T.; Singh, M.; Singh, V. Mitochondrial Dysfunction in Alzheimer’s Disease: Opportunities for Drug Development. Curr. Neuropharmacol. 2022, 20, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Xiao, Q.; Yang, L.; Hu, H.; Ke, Y. Artesunate targets oral tongue squamous cell carcinoma via mitochondrial dysfunction-dependent oxidative damage and Akt/AMPK/mTOR inhibition. J. Bioenerg. Biomembr. 2020, 52, 113–121. [Google Scholar] [CrossRef]
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012, 123, 53–70. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Hajdú, B.; Lőrincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. Fine-tuning of AMPK-ULK1-mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci. Rep. 2020, 10, 17803. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef] [PubMed]
- Brokaw, D.L.; Piras, I.S.; Mastroeni, D.; Weisenberger, D.J.; Nolz, J.; Delvaux, E.; Serrano, G.E.; Beach, T.G.; Huentelman, M.J.; Coleman, P.D. Cell death and survival pathways in Alzheimer’s disease: An integrative hypothesis testing approach utilizing -omic data sets. Neurobiol. Aging 2020, 95, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Long, Z.; Liu, Y.; Luo, M.; Qiu, Y.; Idris, N.F.B.; Song, A.; Wang, K.; He, G. Dihydroartemisinin Ameliorates Decreased Neuroplasticity-Associated Proteins and Excessive Neuronal Apoptosis in APP/PS1 Mice. Curr. Alzheimer Res. 2020, 17, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Xu, J.; Zheng, W. Artemisinin protects PC12 cells against β-amyloid-induced apoptosis through activation of the ERK1/2 signaling pathway. Redox Biol. 2017, 12, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, D.F.; Luo, R.; Wu, Y.; Zhou, H.; Kong, L.L.; Bi, R.; Yao, Y.G. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement. 2018, 14, 215–229. [Google Scholar] [CrossRef]
- Tanaka, H.; Homma, H.; Fujita, K.; Kondo, K.; Yamada, S.; Jin, X.; Waragai, M.; Ohtomo, G.; Iwata, A.; Tagawa, K.; et al. YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat. Commun. 2020, 11, 507. [Google Scholar] [CrossRef]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Balusu, S.; Horré, K.; Thrupp, N.; Craessaerts, K.; Snellinx, A.; Serneels, L.; T’syen, D.; Chrysidou, I.; Arranz, A.M.; Sierksma, A.; et al. MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’s disease. Science 2023, 381, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, X.; Huang, L.; Qian, H.; Liu, W. Mechanism of pyroptosis in neurodegenerative diseases and its therapeutic potential by traditional Chinese medicine. Front. Pharmacol. 2023, 14, 1122104. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Jiang, X.; Wu, M.; Cao, X.; Bao, W.; Zhu, L.Q. Ferroptosis, a Potential Therapeutic Target in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 704298. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P. Antimalarial and anticancer properties of artesunate and other artemisinins: Current development. Monatsh. Chem. 2021, 152, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef]
- Griffiths, J. Grant SGN Synapse pathology in Alzheimer’s disease. Semin. Cell Dev. Biol. 2023, 139, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Hollnagel, J.O.; Elzoheiry, S.; Gorgas, K.; Kins, S.; Beretta, C.A.; Kirsch, J.; Kuhse, J.; Kann, O.; Kiss, E. Early alterations in hippocampal perisomatic GABAergic synapses and network oscillations in a mouse model of Alzheimer’s disease amyloidosis. PLoS ONE 2019, 14, e0209228. [Google Scholar] [CrossRef] [PubMed]
- Meftah, S.; Gan, J. Alzheimer’s disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression. Front. Synaptic Neurosci. 2023, 15, 1129036. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Kiss, E.; Gorgas, K.; Schlicksupp, A.; Groß, D.; Kins, S.; Kirsch, J.; Kuhse, J. Biphasic Alteration of the Inhibitory Synapse Scaffold Protein Gephyrin in Early and Late Stages of an Alzheimer Disease Model. Am. J. Pathol. 2016, 186, 2279–2291. [Google Scholar] [CrossRef] [PubMed]
- Ambrad Giovannetti, E.; Fuhrmann, M. Unsupervised excitation: GABAergic dysfunctions in Alzheimer’s disease. Brain Res. 2019, 1707, 216–226. [Google Scholar] [CrossRef]
- Mori, H.; Yoshino, Y.; Iga, J.I.; Ochi, S.; Funahashi, Y.; Yamazaki, K.; Kumon, H.; Ozaki, Y.; Ueno, S.I. Aberrant Expression of GABA-Related Genes in the Hippocampus of 3xTg-AD Model Mice from the Early to End Stages of Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 94, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honoré, C.; Courtney, M.; Huber, K.V.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins Target GABAA Receptor Signaling and Impair α Cell Identity. Cell 2017, 168, 86–100.e15. [Google Scholar] [CrossRef]
- Kasaragod, V.B.; Hausrat, T.J.; Schaefer, N.; Kuhn, M.; Christensen, N.R.; Tessmer, I.; Maric, H.M.; Madsen, K.L.; Sotriffer, C.; Villmann, C.; et al. Elucidating the Molecular Basis for Inhibitory Neurotransmission Regulation by Artemisinins. Neuron 2019, 101, 673–689.e11. [Google Scholar] [CrossRef] [PubMed]
- Kasaragod, V.B.; Pacios-Michelena, A.; Schaefer, N.; Zheng, F.; Bader, N.; Alzheimer, C.; Villmann, C.; Schindelin, H. Pyridoxal kinase inhibition by artemisinins down-regulates inhibitory neurotransmission. Proc. Natl. Acad. Sci. USA 2020, 117, 33235–33245. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Kins, S.; Gorgas, K.; Orlik, M.; Fischer, C.; Endres, K.; Schlicksupp, A.; Kirsch, J.; Kuhse, J. Artemisinin-treatment in pre-symptomatic APP-PS1 mice increases gephyrin phosphorylation at Ser270: A modification regulating postsynaptic GABAAR density. Biol. Chem. 2021, 403, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Kuhse, J.; Groeneweg, F.; Kins, S.; Gorgas, K.; Nawrotzki, R.; Kirsch, J.; Kiss, E. Loss of Extrasynaptic Inhibitory Glycine Receptors in the Hippocampus of an AD Mouse Model Is Restored by Treatment with Artesunate. Int. J. Mol. Sci. 2023, 24, 4623. [Google Scholar] [CrossRef] [PubMed]
- Poorgholam, P.; Yaghmaei, P.; Noureddini, M.; Hajebrahimi, Z. Artemisin and human endometrial-derived stem cells improve cognitive function and synaptic plasticity in a rat model of Alzheimer disease and diabetes. Metab. Brain Dis. 2023, 38, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Pang, Y.; Li, J.; Wu, B.; Du, Y.; Chen, Y.; Luo, M.; Wang, Y.; Dong, Z. Dihydroartemisinin Induces O-GlcNAcylation and Improves Cognitive Function in a Mouse Model of Tauopathy. J. Alzheimer’s Dis. 2021, 84, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the Adult Hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018812. [Google Scholar] [CrossRef]
- Scopa, C.; Marrocco, F.; Latina, V.; Ruggeri, F.; Corvaglia, V.; La Regina, F.; Ammassari-Teule, M.; Middei, S.; Amadoro, G.; Meli, G.; et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 2020, 27, 934–948. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 2, 554–560. [Google Scholar] [CrossRef]
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87. [Google Scholar] [CrossRef]
- Sasaki, K.; Geribaldi-Doldán, N.; Wu, Q.; Davies, J.; Szele, F.G.; Isoda, H. Microalgae Aurantiochytrium Sp. Increases Neurogenesis and Improves Spatial Learning and Memory in Senescence-Accelerated Mouse-Prone 8 Mice. Front. Cell Dev. Biol. 2021, 8, 600575. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Mitkevich, V.A.; Makarov, A.A. Effect of β-amyloid on blood-brain barrier properties and function. Biophys. Rev. 2023, 15, 183–197. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Ando, K.; Nagaraj, S.; Küçükali, F.; de Fisenne, M.A.; Kosa, A.C.; Doeraene, E.; Lopez Gutierrez, L.; Brion, J.P.; Leroy, K. PICALM and Alzheimer’s Disease: An Update and Perspectives. Cells 2022, 11, 3994. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Lee, L.; Palmeri, A.; Calabrese, G.; Arancio, O. Behavioral assays with mouse models of Alzheimer’s disease: Practical considerations and guidelines. Biochem. Pharmacol. 2014, 88, 450–467. [Google Scholar] [CrossRef]
- Mssusa, A.K.; Fimbo, A.M.; Nkayamba, A.F.; Irunde, H.F.; Sillo, H.B.; Shewiyo, D.H.; Hill, G.; Minzi, O.M. Safety Profile of Artemether-Lumefantrine: A Cohort Event Monitoring Study in Public Health Facilities in Tanzania. Clin. Drug Investig. 2016, 36, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Roussel, C.; Caumes, E.; Thellier, M.; Ndour, P.A.; Buffet, P.A.; Jauréguiberry, S. Artesunate to treat severe malaria in travellers: Review of efficacy and safety and practical implications. J. Travel Med. 2017, 24, taw093. [Google Scholar] [CrossRef]
- Nontprasert, A.; Pukrittayakamee, S.; Nosten-Bertrand, M.; Vanijanonta, S.; White, N.J. Studies of the neurotoxicity of oral artemisinin derivatives in mice. Am. J. Trop. Med. Hyg. 2000, 62, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Wesche, D.L.; DeCoster, M.A.; Tortella, F.C.; Brewer, T.G. Neurotoxicity of artemisinin analogs in vitro. Antimicrob. Agents Chemother. 1994, 38, 1813–1819. [Google Scholar] [CrossRef]
- Singh, S.; Giri, A.; Giri, S. The antimalarial agent artesunate causes sperm DNA damage and hepatic antioxidant defense in mice. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 777, 1–6. [Google Scholar] [CrossRef]
- Zheng, C.; Shan, L.; Tong, P.; Efferth, T. Cardiotoxicity and Cardioprotection by Artesunate in Larval Zebrafish. Dose-Response 2020, 18, 1559. [Google Scholar] [CrossRef] [PubMed]
- Gordi, T.; Lepist, E.I. Artemisinin derivatives: Toxic for laboratory animals, safe for humans? Toxicol. Lett. 2004, 147, 99–107. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Aβ | pTau | Inflammation | ROS | Autophagy | Cell Death | Synapse Proteins | Neurogenesis | BBB | Memory Cognition | Substance | Dose/Day, Route and Duration of Administration/ Concentration | Lowest Efficient Dose/ Concentration | Molecular Mechanisms | System/Start of Therapy | References | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | | | |  | | | | | | - | - | - | - | - | - | |
| | Artemisinin | 40 mg/kg, i.p., 30 days | 40 mg/kg | NF-κB, IL-6, TNF-α | (−) | APPswe/PS1dE9 mice, 4-month-old | Shi et al., (2013) [72] | ||||||||
| NALP3 | (−) | |||||||||||||||
| BACE1 | (−) | |||||||||||||||
| | Artemisinin | 3.1–100 μM | 1.5 μM | ERK1/2 pathway | (+) | PC12 cells + Aβ25–35 | Zeng et al., (2017) [118] | ||||||||
| Caspase-3 and -7 | (−) | |||||||||||||||
| | | Artemisinin B | 20-, 40-, and 80 mg/kg, i.g., 1–2 weeks 1–8 μM | 20 mg/kg 1 μM | TLR4, MyD88, NF-κB | (−) | KM mice- intra-ventricular Aβ25–35 start:immediately after surgery BV2 cells | Qiang et al., (2018) [95] | |||||||
| IL-1β, IL-6, TNF-α | (−) | |||||||||||||||
| | | | | Artemether | 5- and 20 mg/kg, i.p., 4 weeks, 3–100 μM |
5 mg/kg 10 μM | AMPK/GSK3β/Nrf2 | (+) | 3xTg mice, 8-month-old PC12 and SH-SY5Y, primary cortical neurons | Li et al., (2019) [82] | |||||
| Bcl-2 | (+) | |||||||||||||||
| Bax | (−) | |||||||||||||||
| | | | | | Artemisinin | 1-, 5- and 10 mg/kg, i.p., 30 days 3.125–100 μM | 1 mg/kg, 6.25 μM | ERK/CREB pathway | (+) | 3xTg mice, 11-month-old SH-SY5Y cells | Zhao et al., (2020) [84] | ||||
| Cytochrome C | (−) | |||||||||||||||
| Caspase-3 and -9 | (−) | |||||||||||||||
| Bcl-2/Bax | (+) | |||||||||||||||
| | | Dihydro-artemisinin | 20 mg/kg, p.o., 90 days | 20 mg/kg | BACE1 | (−) | APPswe/PSEN1dE9, 6-month-old N2a-APP and SH-SY5Y-APP cells | Zhao et al., (2020) [73] | |||||||
| mTOR/ ULK1 | (+) | |||||||||||||||
| | Dihydro artemisinin | 20 mg/kg. p.o., 90 days | 20 mg/kg | Caspase-3, Bax. | (−) | APPswe/PSEN1dE9, 6-month-old | Zhao et al., (2020) [117] | ||||||||
| Bcl-2 | (+) | |||||||||||||||
| | Artemisinin, Artesunate | 10- and 100 mg/kg, p.o., 3 months 0.05, 0.125, 0.25 μM | 10 mg/kg 0.05 μM | CTFs | (−) | APPswe/PS1L166P, 9-month-old Hippocampal neurons | Kiss et al., (2021) [77] | ||||||||
| Artemisinin | 10- and 100 mg/kg, p.o., 6 weeks | 10 mg/kg | Gephyrin phosphorylation | (+) | APPswe/PS1L166P, 6-week-old | Kiss et al., (2021) [143] | |||||||||
| | | Artemisinin | 50 mg/kg, i.p., 4 weeks | 50 mg/kg | ROS, TNF-α | (−) | Streptozotocin-induced AD and diabetes in rats | Poorgholam et al., (2021) [103] | |||||||
| Blood glucose | (−) | |||||||||||||||
| | | Artemether | n.n. | n.n. | BACE1, mTOR | (−) | Aβ25–35-treated rats N2a cells | Li et al., (2021) [83] | |||||||
| | | | | | Artesunate | 5- and 10 mg/kg, p.p., 6 months 0.01, 0.05, 0.1, 0.2, 0.5, 1.0 μM | 5 mg/kg 0.1 μM | TNFα, IL-6, IL-1β | (−) | APPswe/PS1, 2-month-old BV-2-, N2a cells | Qin et al., (2022) [75] | ||||
| PINK1. | (+) | |||||||||||||||
| Parkin | (+) | |||||||||||||||
| | | | Artemisinin | 5 mg/kg, i.p., 4 weeks 0.25, 0.5, 1.0 μM | 5 mg/kg 0.25 μM | TLR4/NF-κB | (−) | C57 mice injected with Aβ1–42 into the hippocampus, start:4 weeks pre BV2 Cells | Zhao et al., (2022) [96] | ||||||
| TNF-α, IL-1β, IL-6 | (−) | |||||||||||||||
| | | Dihydro-artemisinin | 50- and 300 mg/kg, p.o., 4/ 6 months | 50 mg/kg | - | APPswePSEN1dE9/Nju, 3-month-old | Xiao et al., (2022) [74] | ||||||||
| | | | Artesunate | 32 mg/kg, i.p., 2 months, 3 μM | EC50: 2.1 μM | Brain capillary PICALM | (+) | 5XFAD mice, 3-month-old HEK293t luciferase reporter line | Kisler et al., (2023) [76] | ||||||
| Artesunate | 10- and 100 mg/kg, p.o., 1.5/3 months | 10 mg/kg | - | APPswe/PS1L166P, 1.5-/ 9-month- old | Kuhse et al., (2023) [144] | ||||||||||
| | Artemisinin | 50 mg/kg, i.p., 4 weeks, | 50 mg/kg; | Blood glucose | (−) | Streptozotocin-induced AD and diabetes in rats, 4-month-old | Poorgholam et al., (2023) [145] | ||||||||
| | | | | | | | Artemisia annua extract | 6.7- and 20 mg/mL, p.o., 3 months 1–10,000 μg/mL | 6.7 mg/mL 30 μg/mL | IL-6, TNF-α, IL1β | (−) | 3xTg mice, 9-month-old SH-SY5Y, primary neurons PC12 cells | Zhou et al., (2023) [85] | ||
| Hippo/YAP signaling | (+) | |||||||||||||||
| Bax | (−) | |||||||||||||||
| Bcl-2 | (+) | |||||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss, E.; Kins, S.; Gorgas, K.; Venczel Szakács, K.H.; Kirsch, J.; Kuhse, J. Another Use for a Proven Drug: Experimental Evidence for the Potential of Artemisinin and Its Derivatives to Treat Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 4165. https://doi.org/10.3390/ijms25084165
Kiss E, Kins S, Gorgas K, Venczel Szakács KH, Kirsch J, Kuhse J. Another Use for a Proven Drug: Experimental Evidence for the Potential of Artemisinin and Its Derivatives to Treat Alzheimer’s Disease. International Journal of Molecular Sciences. 2024; 25(8):4165. https://doi.org/10.3390/ijms25084165
Chicago/Turabian StyleKiss, Eva, Stefan Kins, Karin Gorgas, Kinga Hajnal Venczel Szakács, Joachim Kirsch, and Jochen Kuhse. 2024. "Another Use for a Proven Drug: Experimental Evidence for the Potential of Artemisinin and Its Derivatives to Treat Alzheimer’s Disease" International Journal of Molecular Sciences 25, no. 8: 4165. https://doi.org/10.3390/ijms25084165
APA StyleKiss, E., Kins, S., Gorgas, K., Venczel Szakács, K. H., Kirsch, J., & Kuhse, J. (2024). Another Use for a Proven Drug: Experimental Evidence for the Potential of Artemisinin and Its Derivatives to Treat Alzheimer’s Disease. International Journal of Molecular Sciences, 25(8), 4165. https://doi.org/10.3390/ijms25084165