
Regulation of Molecular Biomarkers Associated with the Progression of Prostate Cancer

Abstract
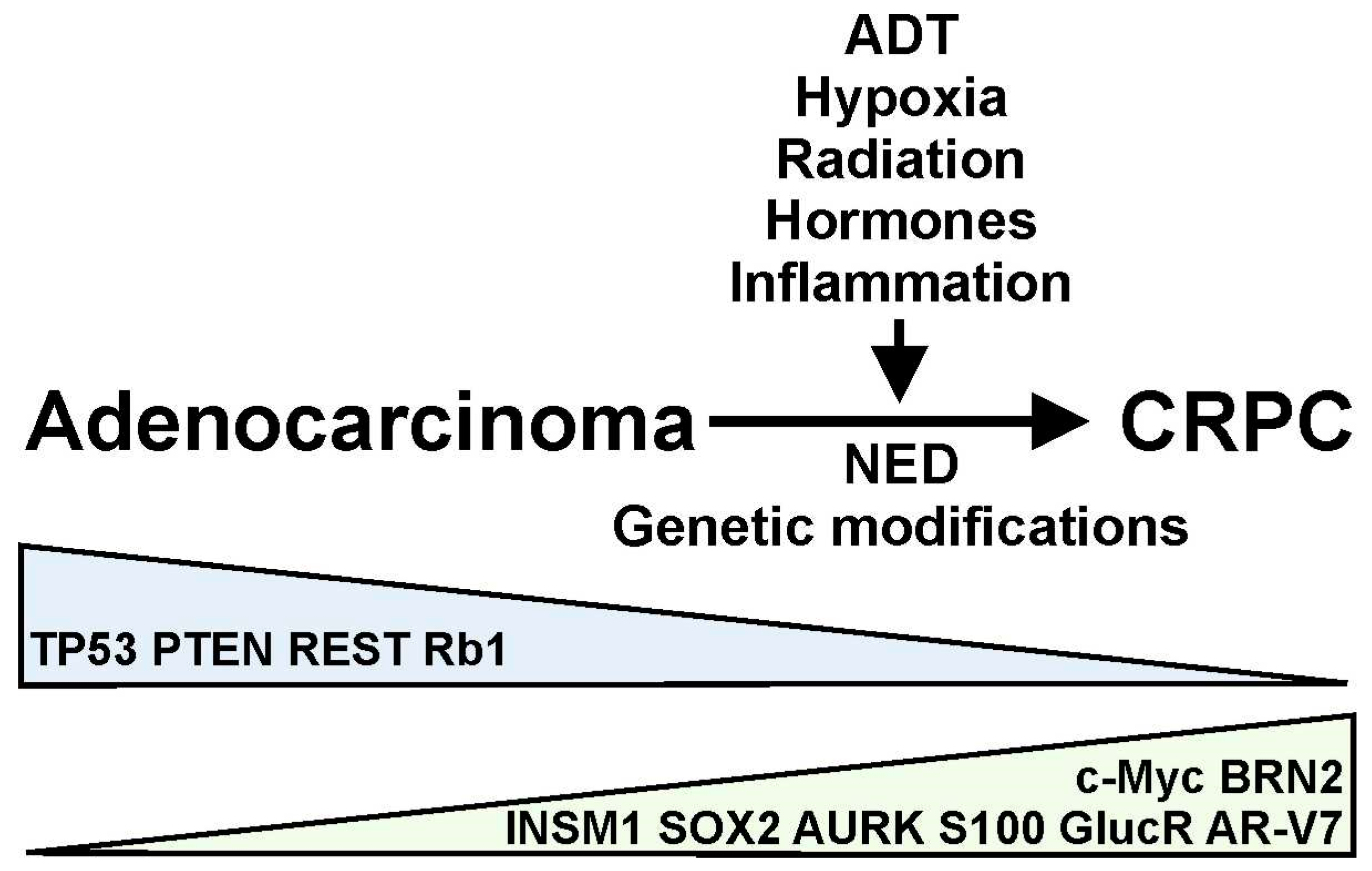
:1. Introduction
2. Cell Signaling Molecules and Receptors: PTEN, Aurora Kinases, Retinoblastoma Tumor Suppressor, Glucocorticoid Receptor, AR-V7, and Others
3. Conclusions
4. Future Directions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADT | androgen deprivation therapy |
| AR | androgen receptor |
| CRPC | castration-resistant prostate cancer |
| DHT | dihydrotestosterone |
| EMT | epithelial-to-mesenchymal transition |
| FBS | fetal bovine serum |
| IL-6 | interleukin-6 |
| LNCaP | lymph node carcinoma of the prostate |
| NE | neuroendocrine |
| NED | neuroendocrine differentiation |
| NSE | neuron-specific enolase |
| PCa | prostate cancer |
| PSA | prostate-specific antigen |
| SCC | small cell carcinoma |
| TME | tumor microenvironment |
| t-NEPC | therapy-induced neuroendocrine prostate cancer |
References
- American Cancer Society. Cancer Facts & Figures 2017; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Crona, D.J.; Whang, Y.E. Androgen receptor-dependent and -independent mechanisms involved in prostate cancer therapy resistance. Cancers 2017, 9, 67. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Bruchovsky, N.; Wilson, J.D. The intranuclear binding of testosterone and 5-alpha-androstan-17-beta-ol-3-one by rat prostate. J. Biol. Chem. 1968, 243, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Page, S.T.; Lin, D.W.; Fazli, L.; Coleman, I.M.; True, L.D.; Knudsen, B.; Hess, D.L.; Nelson, C.C.; Matsumoto, A.M.; et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007, 67, 5033–5041. [Google Scholar] [CrossRef] [PubMed]
- Mizokami, A.; Koh, E.; Fujita, H.; Maeda, Y.; Egawa, M.; Koshida, K.; Honma, S.; Keller, E.T.; Namiki, M. The adrenal androgen androstenediol is present in prostate cancer tissue after androgen deprivation therapy and activates mutated androgen receptor. Cancer Res. 2004, 64, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed]
- Mahon, K.L.; Lin, H.M.; Castillo, L.; Lee, B.Y.; Lee-Ng, M.; Chatfield, M.D.; Chiam, K.; Breit, S.N.; Brown, D.A.; Molloy, M.P.; et al. Cytokine profiling of docetaxel-resistant castration-resistant prostate cancer. Br. J. Cancer. 2015, 112, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. High-Fat Diet-induced inflammation accelerates prostate cancer growth via IL6 signaling. Clin. Cancer Res. 2018, 24, 4309–4318. [Google Scholar] [CrossRef]
- Hu, C.D.; Choo, R.; Huang, J. Neuroendocrine differentiation in prostate cancer: A mechanism of radioresistance and treatment failure. Front. Oncol. 2015, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Beltran, H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front. Oncol. 2014, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.K.; Chugh, N.; Tripathi, M. Neuroendocrine Differentiation of Prostate Cancer-An Intriguing Example of Tumor Evolution at Play. Cancers 2019, 11, 1405. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.; Lu, J.F.; Chen, H.C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef]
- Hirano, D.; Okada, Y.; Minei, S.; Takimoto, Y.; Nemoto, N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur. Urol. 2004, 45, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Ploussard, G.; Allory, Y.; Nicolaiew, N.; Boissière-Michot, F.; Maillé, P.; Kheuang, L.; Coppolani, E.; Ali, A.; Bibeau, F.; et al. Increased expression of class III beta-tubulin in castration-resistant human prostate cancer. Br. J. Cancer. 2009, 101, 951–956. [Google Scholar] [CrossRef]
- Alberti, C. Neuroendocrine differentiation in prostate carcinoma: Focusing on its pathophysiologic mechanisms and pathological features. II G. Chir. J. Ital. Assoc. Hosp. Surg. 2010, 31, 568–574. [Google Scholar]
- Su, R.; Chen, L.; Jiang, Z.; Yu, M.; Zhang, W.; Ma, Z.; Ji, Y.; Shen, K.; Xin, Z.; Qi, J.; et al. Comprehensive analysis of androgen receptor status in prostate cancer with neuroendocrine differentiation. Front. Oncol. 2022, 12, 955166. [Google Scholar] [CrossRef]
- Kaur, H.; Samarska, I.; Lu, J.; Faisal, F.; Maughan, B.L.; Murali, S.; Asrani, K.; Alshalalfa, M.; Antonarakis, E.S.; Epstein, J.I.; et al. Neuroendocrine differentiation in usual-type prostatic adenocarcinoma: Molecular characterization and clinical significance. Prostate 2020, 80, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Bonkhoff, H.; Wernert, N.; Dhom, G.; Remberger, K. Relation of endocrine-paracrine cells to cell proliferation in normal, hyperplastic, and neoplastic human prostate. Prostate 1991, 19, 91–98. [Google Scholar] [CrossRef]
- Abrahamsson, P.A. Neuroendocrine cells in tumour growth of the prostate. Endocr. Relat. Cancer 1999, 6, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Grobholz, R.; Griebe, M.; Sauer, C.G.; Michel, M.S.; Trojan, L.; Bleyl, U. Influence of neuroendocrine tumor cells on proliferation in prostatic carcinoma. Hum. Pathol. 2005, 36, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.L.; Madeb, R.; Bourne, P.; Lei, J.; Yang, X.; Tickoo, S.; Liu, Z.; Tan, D.; Cheng, L.; Hatem, F.; et al. Small cell carcinoma of the prostate: An immunohistochemical study. Am. J. Surg. Pathol. 2006, 30, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Miao, J.; Wang, Y.; Luo, W.; Ji, Z.; Lai, H.; Zhang, M.; Cheng, X.; Wang, J.; Fang, Y.; et al. Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun. Biol. 2020, 3, 778. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Pirnia, F.; Fang, W.G.; Kang, W.K.; Sartor, O.; Whitesell, L.; Ha, M.J.; Tsokos, M.; Sheahan, M.D.; Nguyen, P.; et al. Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AM.P. Proc. Natl. Acad. Sci. USA 1994, 91, 5330–5334. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Dorai, T.; Szaboles, M.; Katz, A.E.; Olsson, C.A.; Buttyan, R. Transdifferentiation of cultured human prostate cancer cells to a neuroendocrine cell phenotype in a hormone-depleted medium. Urol. Oncol. 1997, 3, 67–75. [Google Scholar] [CrossRef]
- Deeble, P.D.; Murphy, D.J.; Parsons, S.J.; Cox, M.E. Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol. Cell. Biol. 2001, 21, 8471–8482. [Google Scholar] [CrossRef]
- Zelivianski, S.; Verni, M.; Moore, C.; Kondrikov, D.; Taylor, R.; Lin, M.F. Multipathways for transdifferentiation of human prostate cancer cells into neuroendocrine-like phenotype. Biochim. Biophys. Acta. 2001, 1539, 28–43. [Google Scholar]
- Danza, G.; Di Serio, C.; Rosati, F.; Lonetto, G.; Sturli, N.; Kacer, D.; Pennella, A.; Ventimiglia, G.; Barucci, R.; Piscazzi, A.; et al. Notch signaling modulates hypoxia-induced neuroendocrine differentiation of human prostate cancer cells. Mol. Cancer Res. 2012, 10, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Elzey, B.D.; Poulson, J.M.; Morrison, W.B.; Ko, S.C.; Hahn, N.M.; Ratliff, T.L.; Hu, C.D. Ionizing radiation induces neuroendocrine differentiation of prostate cancer cells in vitro, in vivo and in prostate cancer patients. Am. J. Cancer Res. 2011, 1, 834–844. [Google Scholar]
- Fiandalo, M.V.; Wilton, J.H.; Mantione, K.M.; Wrzosek, C.; Attwood, K.M.; Wu, Y.; Mohler, J.L. Serum-free complete medium, an alternative medium to mimic androgen deprivation in human prostate cancer cell line models. Prostate 2018, 78, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Collado, B.; Gutiérrez-Cañas, I.; Rodríguez-Henche, N.; Prieto, J.C.; Carmena, M.J. Vasoactive intestinal peptide increases vascular endothelial growth factor expression and neuroendocrine differentiation in human prostate cancer LNCaP cells. Regul. Pept. 2004, 119, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Urbanucci, A.; Nielsen, H.K.; Guldvik, I.J.; Engedal, A.; Ketola, K.; Wang, W.; Svindland, A.; et al. The β2-Adrenergic Receptor Is a Molecular Switch for Neuroendocrine Transdifferentiation of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 2154–2168. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, W. Beta-adrenergic signaling on neuroendocrine differentiation, angiogenesis, and metastasis in prostate cancer progression. Asian J. Androl. 2019, 21, 253–259. [Google Scholar]
- Hobisch, A.; Rogatsch, H.; Hittmair, A.; Fuchs, D.; Bartsch, G., Jr.; Klocker, H.; Bartsch, G.; Culig, Z. Immunohistochemical localization of interleukin-6 and its receptor in benign, premalignant and malignant prostate tissue. J. Pathol. 2000, 191, 239–244. [Google Scholar] [CrossRef]
- Movsas, B.; Chapman, J.D.; Greenberg, R.E.; Hanlon, A.L.; Horwitz, E.M.; Pinover, W.H.; Stobbe, C.; Hanks, G.E. Increasing levels of hypoxia in prostate carcinoma correlate significantly with increasing clinical stage and patient age: An Eppendorf pO2 study. Cancer 2000, 89, 2018–2024. [Google Scholar] [CrossRef]
- Drachenberg, D.E.; Elgamal, A.A.; Rowbotham, R.; Peterson, M.; Murphy, G.P. Circulating levels of interleukin-6 in patients with hormone refractory prostate cancer. Prostate 1999, 41, 127–133. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer. 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Wu, M.; Wang, X.; McGregor, N.; Pienta, K.J.; Zhang, J. Dynamic regulation of Rad51 by E2F1 and p53 in prostate cancer cells upon drug-induced DNA damage under hypoxia. Mol. Pharmacol. 2014, 85, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.; Wu, J. Differential expression and regulation of p53 in human prostatic cells. Int. J. Oncol. 1997, 10, 1109–1112. [Google Scholar] [CrossRef]
- Liang, H.; Studach, L.; Hullinger, R.L.; Xie, J.; Andrisani, O.M. Down-regulation of RE-1 silencing transcription factor (REST) in advanced prostate cancer by hypoxia-induced miR-106b~25. Exp. Cell Res. 2014, 320, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Cui, Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 induces neuroendocrine differentiation (NED) through suppression of RE-1 silencing transcription factor (REST). Prostate 2014, 74, 1086–1094. [Google Scholar] [CrossRef]
- Bennett, J.L.; Jackson, B.N.; Miller, R.J.; Tsui, H.; Martin-Caraballo, M. IL-6 evoked biochemical changes in prostate cancer cells. Cytokine 2023, 161, 156079. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.P.; Chang, Y.T.; Lee, S.Y.; Campbell, M.; Wang, T.C.; Shen, S.H.; Chung, H.J.; Chang, Y.H.; Chiu, A.W.; Pan, C.C.; et al. REST reduction is essential for hypoxia-induced neuroendocrine differentiation of prostate cancer cells by activating autophagy signaling. Oncotarget 2016, 7, 26137–26151. [Google Scholar] [CrossRef]
- Chen, R.; Li, Y.; Buttyan, R.; Dong, X. Implications of PI3K/AKT inhibition on REST protein stability and neuroendocrine phenotype acquisition in prostate cancer cells. Oncotarget 2017, 8, 84863–84876. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.G.; Cho, H.; Herzka, T.; Watrud, K.; DeMarco, D.V.; Wang, V.M.; Senturk, S.; Fellmann, C.; Ding, D.; Beinortas, T.; et al. MYC Drives Pten/Trp53-Deficient Proliferation and Metastasis due to IL6 Secretion and AKT Suppression via PHLPP2. Cancer Discov. 2015, 5, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Chung, T.D. STAT3 mediates IL-6-induced growth inhibition in the human prostate cancer cell line LNCaP. Prostate 2000, 42, 88–98. [Google Scholar] [CrossRef]
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H. c-MYC drives histone demethylase PHF8 during neuroendocrine differentiation and in castration-resistant prostate cancer. Oncotarget 2016, 7, 75585–75602. [Google Scholar] [CrossRef]
- Monga, J.; Adrianto, I.; Rogers, C.; Gadgeel, S.; Chitale, D.; Alumkal, J.J.; Beltran, H.; Zoubeidi, A.; Ghosh, J. Tribbles 2 pseudokinase confers enzalutamide resistance in prostate cancer by promoting lineage plasticity. J. Biol. Chem. 2022, 298, 101556. [Google Scholar] [CrossRef]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer. 2009, 9, 874–885. [Google Scholar] [CrossRef]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S.; et al. MUC1-C regulates lineage plasticity driving progression to neuroendocrine prostate cancer. Nat. Commun. 2020, 11, 338. [Google Scholar] [CrossRef]
- Kregel, S.; Kiriluk, K.J.; Rosen, A.M.; Cai, Y.; Reyes, E.E.; Otto, K.B.; Tom, W.; Paner, G.P.; Szmulewitz, R.Z.; Vander Griend, D.J. Sox2 is an androgen receptor-repressed gene that promotes castra-tion-resistant prostate cancer. PLoS ONE 2013, 8, e53701. [Google Scholar] [CrossRef]
- Yu, X.; Cates, J.M.; Morrissey, C.; You, C.; Grabowska, M.M.; Zhang, J.; DeGraff, D.J.; Strand, D.W.; Franco, O.E.; Lin-Tsai, O.; et al. SOX2 expression in the developing, adult, as well as, diseased prostate. Prostate Cancer Prostatic Dis. 2014, 17, 301–309. [Google Scholar] [CrossRef]
- Russo, M.V.; Esposito, S.; Tupone, M.G.; Manzoli, L.; Airoldi, I.; Pompa, P.; Cindolo, L.; Schips, L.; Sorrentino, C.; Di Carlo, E. SOX2 boosts major tumor progression genes in prostate cancer and is a functional biomarker of lymph node metastasis. Oncotarget 2016, 7, 12372–12385. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Nan, B.; Snabboon, T.; Unni, E.; Yuan, X.-J.; Whang, Y.E.; Marcelli, M. The PTEN tumor suppressor is a negative modulator of androgen receptor transcriptional activity. J. Mol. Endocrinol. 2003, 31, 169–183. [Google Scholar] [CrossRef]
- Gujrati, H.; Ha, S.; Wang, B.D. Deregulated microRNAs involved in prostate cancer aggressiveness and treatment resistance mechanisms. Cancers 2023, 15, 3140. [Google Scholar] [CrossRef]
- Kivinummi, K.; Urbanucci, A.; Leinonen, K.; Tammela, T.L.J.; Annala, M.; Isaacs, W.B.; Bova, G.S.; Nykter, M.; Visakorpi, T. The expression of AURKA is androgen regulated in castration-resistant prostate cancer. Sci. Rep. 2017, 7, 17978. [Google Scholar] [CrossRef]
- Yang, S.; He, S.; Zhou, X.; Liu, M.; Zhu, H.; Wang, Y.; Zhang, W.; Yan, S.; Quan, L.; Bai, J.; et al. Suppression of Aurora-A oncogenic potential by c-Myc downregulation. Exp. Mol. Med. 2010, 42, 759–767. [Google Scholar] [CrossRef]
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 2014, 42, 999–1015. [Google Scholar] [CrossRef]
- Hong, S.K.; Kim, J.H.; Lin, M.F.; Park, J.I. The Raf/MEK/extracellular signal-regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp. Cell Res. 2011, 317, 2671–2682. [Google Scholar] [CrossRef]
- Eder, I.E.; Haag, P.; Basik, M.; Mousses, S.; Bektic, J.; Bartsch, G.; Klocker, H. Gene expression changes following androgen receptor elimination in LNCaP prostate cancer cells. Mol. Carcinog. 2003, 37, 181–191. [Google Scholar] [CrossRef]
- Bernichtein, S.; Pigat, N.; Barry Delongchamps, N.; Boutillon, F.; Verkarre, V.; Camparo, P.; Reyes-Gomez, E.; Méjean, A.; Oudard, S.M.; Lepicard, E.M.; et al. Vitamin D3 Prevents Calcium-Induced Progression of Early-Stage Prostate Tumors by Counteracting TRPC6 and Calcium Sensing Receptor Upregulation. Cancer Res. 2017, 77, 355–365. [Google Scholar] [CrossRef]
- Xie, N.; Cheng, H.; Lin, D.; Liu, L.; Yang, O.; Jia, L.; Fazli, L.; Gleave, M.E.; Wang, Y.; Rennie, P.; et al. The expression of glucocorticoid receptor is negatively regulated by active androgen receptor signaling in prostate tumors. Int. J. Cancer 2015, 136, E27–E38. [Google Scholar] [CrossRef]
- Mediwala, S.N.; Sun, H.; Szafran, A.T.; Hartig, S.M.; Sonpavde, G.; Hayes, T.G.; Thiagarajan, P.; Mancini, M.A.; Marcelli, M. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate 2013, 73, 267–277. [Google Scholar] [CrossRef]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef]
- Fan, L.; Xu, S.; Zhang, F.; Cui, X.; Fazli, L.; Gleave, M.; Clark, D.J.; Yang, A.; Hussain, A.; Rassool, F.; et al. Histone demethylase JMJD1A promotes expression of DNA repair factors and radio-resistance of prostate cancer cells. Cell Death Dis. 2020, 11, 214. [Google Scholar] [CrossRef]
- Li, Y.; Xie, N.; Gleave, M.E.; Rennie, P.S.; Dong, X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget 2015, 6, 33743–33754. [Google Scholar] [CrossRef]
- Kiliccioglu, I.; Konac, E.; Dikmen, A.U.; Sozen, S.; Bilen, C.Y. Hsp-27 and NF-κB pathway is associated with AR/AR-V7 expression in prostate cancer cells. Gene 2019, 697, 138–143. [Google Scholar] [CrossRef]
- Morel, K.L.; Hamid, A.A.; Clohessy, J.G.; Pandell, N.; Ellis, L.; Sweeney, C.J. NF-κB Blockade with Oral Administration of Dimethylaminoparthenolide (DMAPT), Delays Prostate Cancer Resistance to Androgen Receptor (AR) Inhibition and Inhibits AR Variants. Mol. Cancer Res. 2021, 19, 1137–1145. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769. [Google Scholar] [CrossRef]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef]
- Hamid, A.A.; Gray, K.P.; Shaw, G.; MacConaill, L.E.; Evan, C.; Bernard, B.; Loda, M.; Corcoran, N.M.; Van Allen, E.M.; Choudhury, A.D.; et al. Compound genomic alterations of TP53, PT.E.N.; and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur. Urol. 2019, 76, 89–97. [Google Scholar] [CrossRef]
- De Laere, B.; Oeyen, S.; Mayrhofer, M.; Whitington, T.; van Dam, P.-J.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; et al. TP53 Outperforms Other Androgen Receptor Biomarkers to Predict Abiraterone or Enzalutamide Outcome in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 1766–1773. [Google Scholar] [CrossRef]
- Liu, C.; Zhu, Y.; Lou, W.; Nadiminty, N.; Chen, X.; Zhou, Q.; Shi, X.B.; deVere White, R.W.; Gao, A.C. Functional p53 determines docetaxel sensitivity in prostate cancer cells. Prostate 2013, 73, 418–427. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef]
- Chopra, H.; Khan, Z.; Contreras, J.; Wang, H.; Sedrak, A.; Zhu, Y. Activation of p53 and destabilization of androgen receptor by combinatorial inhibition of MDM2 and MDMX in prostate cancer cells. Oncotarget 2018, 9, 6270–6281. [Google Scholar] [CrossRef]
- Brown, D.W.; Beatty, P.H.; Lewis, J.D. Molecular targeting of the most functionally complex gene in precision oncology: p53. Cancers 2022, 14, 5176. [Google Scholar] [CrossRef]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef]
- Lapuk, A.V.; Wu, C.; Wyatt, A.W.; McPherson, A.; McConeghy, B.J.; Brahmbhatt, S.; Mo, F.; Zoubeidi, A.; Anderson, S.; Bell, R.H.; et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J. Pathol. 2012, 227, 286–297. [Google Scholar] [CrossRef]
- Flores-Morales, A.; Bergmann, T.B.; Lavallee, C.; Batth, T.S.; Lin, D.; Lerdrup, M.; Friis, S.; Bartels, A.; Kristensen, G.; Krzyzanowska, A.; et al. Proteogenomic characterization of patient-derived xenografts highlights the role of REST in neuroendocrine differentiation of castration-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 595–608. [Google Scholar] [CrossRef]
- Indo, S.; Orellana-Serradell, O.; Torres, M.J.; Castellón, E.A.; Contreras, H.R. Overexpression of REST Represses the Epithelial-Mesenchymal Transition Process and Decreases the Aggressiveness of Prostate Cancer Cells. Int. J. Mol. Sci. 2024, 25, 3332. [Google Scholar] [CrossRef]
- Li, W.; Zheng, D.; Zhang, Y.; Yang, S.; Su, N.; Bakhoum, M.; Zhang, G.; Naderinezhad, S.; Mao, Z.; Wang, Z.; et al. Androgen deprivation induces neuroendocrine phenotypes in prostate cancer cells through CREB1/EZH2-mediated downregulation of REST. Res. Sq. 2023. [CrossRef]
- Henriksson, M.; Lüscher, B. Proteins of the Myc network: Essential regulators of cell growth and differentiation. Adv. Cancer Res. 1996, 68, 109–182. [Google Scholar]
- Quarmby, V.E.; Beckman, W.C., Jr.; Wilson, E.M.; French, F.S. Androgen regulation of c-myc messenger ribonucleic acid levels in rat ventral prostate. Mol. Endocrinol. 1987, 1, 865–874. [Google Scholar] [CrossRef]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef]
- Gao, L.; Schwartzman, J.; Gibbs, A.; Lisac, R.; Kleinschmidt, R.; Wilmot, B.; Bottomly, D.; Coleman, I.; Nelson, P.; McWeeney, S.; et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS ONE 2013, 8, e63563. [Google Scholar] [CrossRef]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Mosquera, J.M.; Beltran, H.; Park, K.; MacDonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.J.; Chen, C.J.; Lin, T.Y.; Liu, Y.Y.; Tseng, L.L.; Cheng, M.L.; Chuu, C.P.; Tsai, H.K.; Kuo, W.L.; Kung, H.J.; et al. Targeting KDM4B that coactivates c-Myc-regulated metabolism to suppress tumor growth in castration-resistant prostate cancer. Theranostics 2021, 11, 7779–7796. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Davies, A.; Ketola, K.; Zoubeidi, A. Regulation of tumor cell plasticity by the androgen receptor in prostate cancer. Endocr.-Relat. Cancer 2015, 22, R165–R182. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Drolet, D.W.; Rosenfeld, M.G. POU-domain proteins: Structure and function of developmental regulators. Curr. Opin. Cell Biol. 1993, 5, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Lan, M.S.; Breslin, M.B. Structure, expression, and biological function of INSM1 transcription factor in neuroendocrine differentiation. FASEB J. 2009, 23, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Romanel, A.; Conteduca, V.; Casiraghi, N.; Sigouros, M.; Franceschini, G.M.; Orlando, F.; Fedrizzi, T.; Ku, S.Y.; Dann, E.; et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J. Clin. Investig. 2020, 130, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, W.D.; Saunee, N.A.; Breslin, M.B.; Lan, M.S. Zinc finger transcription factor INSM1 interrupts cyclin D1 and CDK4 binding and induces cell cycle arrest. J. Biol. Chem. 2009, 284, 5574–5581. [Google Scholar] [CrossRef]
- Xin, Z.; Zhang, Y.; Jiang, Z.; Zhao, L.; Fan, L.; Wang, Y.; Xie, S.; Shangguan, X.; Zhu, Y.; Pan, J.; et al. Insulinoma-associated protein 1 is a novel sensitive and specific marker for small cell carcinoma of the prostate. Hum. Pathol. 2018, 79, 151–159. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef]
- Lovnicki, J.; Gan, Y.; Feng, T.; Li, Y.; Xie, N.; Ho, C.H.; Lee, A.R.; Chen, X.; Nappi, L.; Han, B.; et al. LIN28B promotes the development of neuroendocrine prostate cancer. J. Clin. Investig. 2020, 130, 5338–5348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, T.; Hong, D.; Dong, B.; Wang, Y.; Huang, H.; Zhang, W.; Lian, B.; Ji, B.; Shi, H.; et al. Single-cell transcriptional regulation and genetic evolution of neuroendocrine prostate cancer. iScience 2022, 25, 104576. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signaling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cheville, J.C.; Pan, Y.; Roche, P.C.; Schmidt, L.J.; Tindall, D.J. PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J. Biol. Chem. 2001, 276, 38830–38836. [Google Scholar] [CrossRef]
- Maughan, B.L.; Guedes, L.B.; Boucher, K.; Rajoria, G.; Liu, Z.; Klimek, S.; Zoino, R.; Antonarakis, E.S.; Lotan, T.L. p53 status in the primary tumor predicts efficacy of subsequent abiraterone and enzalutamide in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Feng, Y.; Zhang, C.; Cheng, D.; Wu, R.; Yang, Y.; Sargsyan, D.; Kumar, D.; Kong, A.N. PTEN deletion drives aberrations of DNA methylome and transcriptome in different stages of prostate cancer. ASEB J. 2020, 34, 1304–1318. [Google Scholar] [CrossRef]
- Shukla, S.; Maclennan, G.T.; Hartman, D.J.; Fu, P.; Resnick, M.I.; Gupta, S. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int. J. Cancer 2007, 121, 1424–1432. [Google Scholar] [CrossRef]
- Kollareddy, M.; Dzubak, P.; Zheleva, D.; Hajduch, M. Aurora kinases: Structure, functions and their association with cancer. Biomed. Pap. Med. Fac. Palacky. Univ. Olomouc Czech Repub. 2008, 152, 27–33. [Google Scholar] [CrossRef]
- Lee, E.C.; Frolov, A.; Li, R.; Ayala, G.; Greenberg, N.M. Targeting Aurora kinases for the treatment of prostate cancer. Prostate 2003, 55, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Chieffi, P.; Cozzolino, L.; Kisslinger, A.; Libertini, S.; Staibano, S.; Mansueto, G.; De Rosa, G.; Villacci, A.; Vitale, M.; Linardopoulos, S.; et al. Aurora B expression directly correlates with prostate cancer malignancy and influence prostate cell proliferation. Prostate 2006, 66, 326–333. [Google Scholar] [CrossRef]
- Sun, F.; Zhang, Z.W.; Tan, E.M.; Lim, Z.L.R.; Li, Y.; Wang, X.C.; Chua, S.E.; Li, J.; Cheung, E.; Yong, E.L. Icaritin suppresses development of neuroendocrine differentiation of prostate cancer through inhibition of IL-6/STAT3 and Aurora kinase A pathways in TRAMP mice. Carcinogenesis 2016, 37, 701–711. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhang, M.G.; Wang, X.J.; Zhong, S.; Shao, Y.; Zhu, Y.; Shen, Z.J. AURKA suppression induces DU145 apoptosis and sensitizes DU145 to docetaxel treatment. Am. J. Transl. Res. 2013, 5, 359–367. [Google Scholar] [PubMed]
- Sicotte, H.; Kalari, K.R.; Qin, S.; Dehm, S.M.; Bhargava, V.; Gormley, M.; Tan, W.; Sinnwell, J.P.; Hillman, D.W.; Li, Y.; et al. Molecular Profile Changes in Patients with Castrate-Resistant Prostate Cancer Pre- and Post-Abiraterone/Prednisone Treatment. Mol. Cancer Res. 2022, 20, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Sood, A.; Rahimi, H.A.; Wang, W.; Gupta, N.; Hicks, J.; Mosier, S.; Gocke, C.D.; Epstein, J.I.; Netto, G.J.; et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res. 2014, 20, 890–903. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Sharma, A.; Comstock, C.E.; Knudsen, E.S.; Cao, K.H.; Hess-Wilson, J.K.; Morey, L.M.; Barrera, J.; Knudsen, K.E. Retinoblastoma tumor suppressor status is a critical determinant of therapeutic response in prostate cancer cells. Cancer Res. 2007, 7, 6192–6203. [Google Scholar] [CrossRef]
- Aparicio, A.; Logothetis, C.J.; Maity, S.N. Understanding the lethal variant of prostate cancer: Power of examining extremes. Cancer Discov. 2011, 1, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, N.; Reig, Ò.; Marín-Aguilera, M.; Aversa, C.; Ferrer-Mileo, L.; Font, A.; Rodriguez-Vida, A.; Climent, M.Á.; Cros, S.; Chirivella, I.; et al. Transcriptional Profile Associated with Clinical Outcomes in Metastatic Hormone-Sensitive Prostate Cancer Treated with Androgen Deprivation and Docetaxel. Cancers 2022, 14, 4757. [Google Scholar] [CrossRef] [PubMed]
- Brennen, W.N.; Zhu, Y.; Coleman, I.M.; Dalrymple, S.L.; Antony, L.; Patel, R.A.; Hanratty, B.; Chikarmane, R.; Meeker, A.K.; Zheng, S.L.; et al. Resistance to androgen receptor signaling inhibition does not necessitate development of neuroendocrine prostate cancer. JCI Insight 2021, 6, e146827. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Botelho, H.M.; Morozova-Roche, L.A.; Gomes, C.M. Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 2010, 277, 4578–4590. [Google Scholar] [CrossRef] [PubMed]
- Hermani, A.; De Servi, B.; Medunjanin, S.; Tessier, P.A.; Mayer, D. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp. Cell Res. 2006, 312, 184–197. [Google Scholar] [CrossRef]
- Romanuik, T.L.; Wang, G.; Morozova, O.; Delaney, A.; Marra, M.A.; Sadar, M.D. LNCaP Atlas: Gene expression associated with in vivo progression to castration-recurrent prostate cancer. BMC Med. Genom. 2010, 3, 43. [Google Scholar] [CrossRef]
- Mousses, S.; Bubendorf, L.; Wagner, U.; Hostetter, G.; Kononen, J.; Cornelison, R.; Goldberger, N.; Elkahloun, A.G.; Willi, N.; Koivisto, P.; et al. Clinical validation of candidate genes associated with prostate cancer progression in the CWR22 model system using tissue microarrays. Cancer Res. 2002, 62, 1256–1260. [Google Scholar]
- Hammacher, A.; Thompson, E.W.; Williams, E.D. Interleukin-6 is a potent inducer of S100P.; which is up-regulated in androgen-refractory and metastatic prostate cancer. Int. J. Biochem. Cell Biol. 2005, 37, 442–450. [Google Scholar] [CrossRef]
- Tennakoon, S.; Aggarwal, A.; Kállay, E. The calcium-sensing receptor and the hallmarks of cancer. Biochim. Biophys. Acta. 2016, 1863 Pt B, 1398–1407. [Google Scholar] [CrossRef]
- Ahearn, T.U.; Tchrakian, N.; Wilson, K.M.; Lis, R.; Nuttall, E.; Sesso, H.D.; Loda, M.; Giovannucci, E.; Mucci, L.A.; Finn, S.; et al. Calcium-Sensing Receptor Tumor Expression and Lethal Prostate Cancer Progression. J. Clin. Endocrinol. Metab. 2016, 101, 2520–2527. [Google Scholar] [CrossRef]
- Feng, J.; Xu, X.; Li, B.; Brown, E.; Farris, A.B.; Sun, S.Y.; Yang, J.J. Prostate cancer metastatic to bone has higher expression of the calcium-sensing receptor (CaSR) than primary prostate cancer. Recept. Clin. Investig. 2014, 1, e270. [Google Scholar]
- Sanders, J.L.; Chattopadhyay, N.; Kifor, O.; Yamaguchi, T.; Brown, E.M. Ca2+-sensing receptor expression and PTHrP secretion in PC-3 human prostate cancer cells. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1267–E1274. [Google Scholar] [CrossRef] [PubMed]
- Bery, F.; Cancel, M.; Chantôme, A.; Guibon, R.; Bruyère, F.; Rozet, F.; Mahéo, K.; Fromont, G. The calcium-sensing receptor is a marker and potential driver of neuroendocrine differentiation in prostate cancer. Cancers 2020, 12, 860. [Google Scholar] [CrossRef] [PubMed]
- Ning, P.; Zhong, J.G.; Jiang, F.; Zhang, Y.; Zhao, J.; Tian, F.; Li, W. Role of protein S in castration-resistant prostate cancer-like cells. Endocr. Relat. Cancer 2016, 23, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Musrap, N.; Cretu, D.; Karagiannis, G.S.; Batruch, I.; Smith, C.; Drabovich, A.P.; Trudel, D.; van der Kwast, T.; Morrissey, C.; et al. Proteomic profiling of androgen-independent prostate cancer cell lines reveals a role for protein S during the development of high grade and castration-resistant prostate cancer. J. Biol. Chem. 2012, 287, 34019–34031. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed]
- Pak, S.; Suh, J.; Park, S.Y.; Kim, Y.; Cho, Y.M.; Ahn, H. Glucocorticoid receptor and androgen receptor-targeting therapy in patients with castration-resistant prostate cancer. Front. Oncol. 2022, 12, 972572. [Google Scholar] [CrossRef]
- Kroon, J.; Puhr, M.; Buijs, J.T.; van der Horst, G.; Hemmer, D.M.; Marijt, K.A.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr. Relat. Cancer 2016, 23, 35–45. [Google Scholar] [CrossRef]
- Chen, X.; Chen, F.; Ren, Y.; Weng, G.; Keng, P.C.; Chen, Y.; Lee, S.O. Glucocorticoid receptor upregulation increases radioresistance and triggers androgen independence of prostate cancer. Prostate 2019, 79, 1386–1398. [Google Scholar] [CrossRef]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, L.; Yang, G.; Geng, C.; Luo, Y.; Wu, W.; Manyam, G.C.; Korentzelos, D.; Park, S.; Tang, Z.; et al. PARP inhibition suppresses GR-MYCN-CDK5-RB1-E2F1 signaling and neuroendocrine differentiation in castration-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 6839–6851. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed]
- Tepper, C.G.; Boucher, D.L.; Ryan, P.E.; Ma, A.H.; Xia, L.; Lee, L.F.; Pretlow, T.G.; Kung, H.J. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002, 62, 6606–6614. [Google Scholar]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef]


{kind=link}
| Biomarker | Function | Regulatory Signaling | References |
|---|---|---|---|
| TP53 | Transcription factor, tumor suppressor | Cellular stress, ARS | [41,42,43] |
| RE1-silencing transcription factor (REST) | Transcription factor, neuronal suppressor | IL-6, hypoxia, ARS, PI3K/Akt | [44,45,46,47,48] |
| c-Myc (MYC) | Basic helix–loop–helix zipper transcription factor | IL-6, loss of function of PTEN and TP53 | [49,50,51] |
| BRN2 | POU domain-containing transcription factor | Pseudo-kinase Tribbles 2, Mucin-1 | [52,53,54] |
| Insulinoma-associated protein 1 (INSM1) | Zinc-finger transcription factor, transcriptional repressor | UNK | |
| SOX2 | SRY (sex-determining region Y) box 2 transcription factor | ARS, TP53 | [55,56,57,58] |
| Tensin homolog (PTEN) | PI3K phosphatase | ARS, miRNA | [59,60] |
| Aurora kinases (AURKs) | Serine/threonine kinase | ARS, c-Myc, REST | [61,62,63] |
| Retinoblastoma tumor suppressor protein (Rb1) | Cell cycle regulator, tumor suppressor | IL-6, ARS | [46,64] |
| S100 proteins | Secreted proteins | ARS | [29,46,65] |
| Calcium-sensing receptor (CasR) | G-protein-coupled receptor | VitD | [66] |
| Protein S (also known as PROS) | Plasma glycoprotein | UNK | |
| Glucocorticoid receptor (GlucR) | Intracellular receptor | ARS, cAMP | [46,67] |
| Androgen receptor variant 7 (AR-V7) | Intracellular receptor | ARS, PI3K/Akt, NF-κB | [68,69,70,71,72,73] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin-Caraballo, M. Regulation of Molecular Biomarkers Associated with the Progression of Prostate Cancer. Int. J. Mol. Sci. 2024, 25, 4171. https://doi.org/10.3390/ijms25084171
Martin-Caraballo M. Regulation of Molecular Biomarkers Associated with the Progression of Prostate Cancer. International Journal of Molecular Sciences. 2024; 25(8):4171. https://doi.org/10.3390/ijms25084171
Chicago/Turabian StyleMartin-Caraballo, Miguel. 2024. "Regulation of Molecular Biomarkers Associated with the Progression of Prostate Cancer" International Journal of Molecular Sciences 25, no. 8: 4171. https://doi.org/10.3390/ijms25084171
APA StyleMartin-Caraballo, M. (2024). Regulation of Molecular Biomarkers Associated with the Progression of Prostate Cancer. International Journal of Molecular Sciences, 25(8), 4171. https://doi.org/10.3390/ijms25084171