

Polyploidy Promotes Hypertranscription, Apoptosis Resistance, and Ciliogenesis in Cancer Cells and Mesenchymal Stem Cells of Various Origins: Comparative Transcriptome In Silico Study


Abstract
:1. Introduction
2. Results
2.1. Characterization of Ploidy Regulated Gene Sets
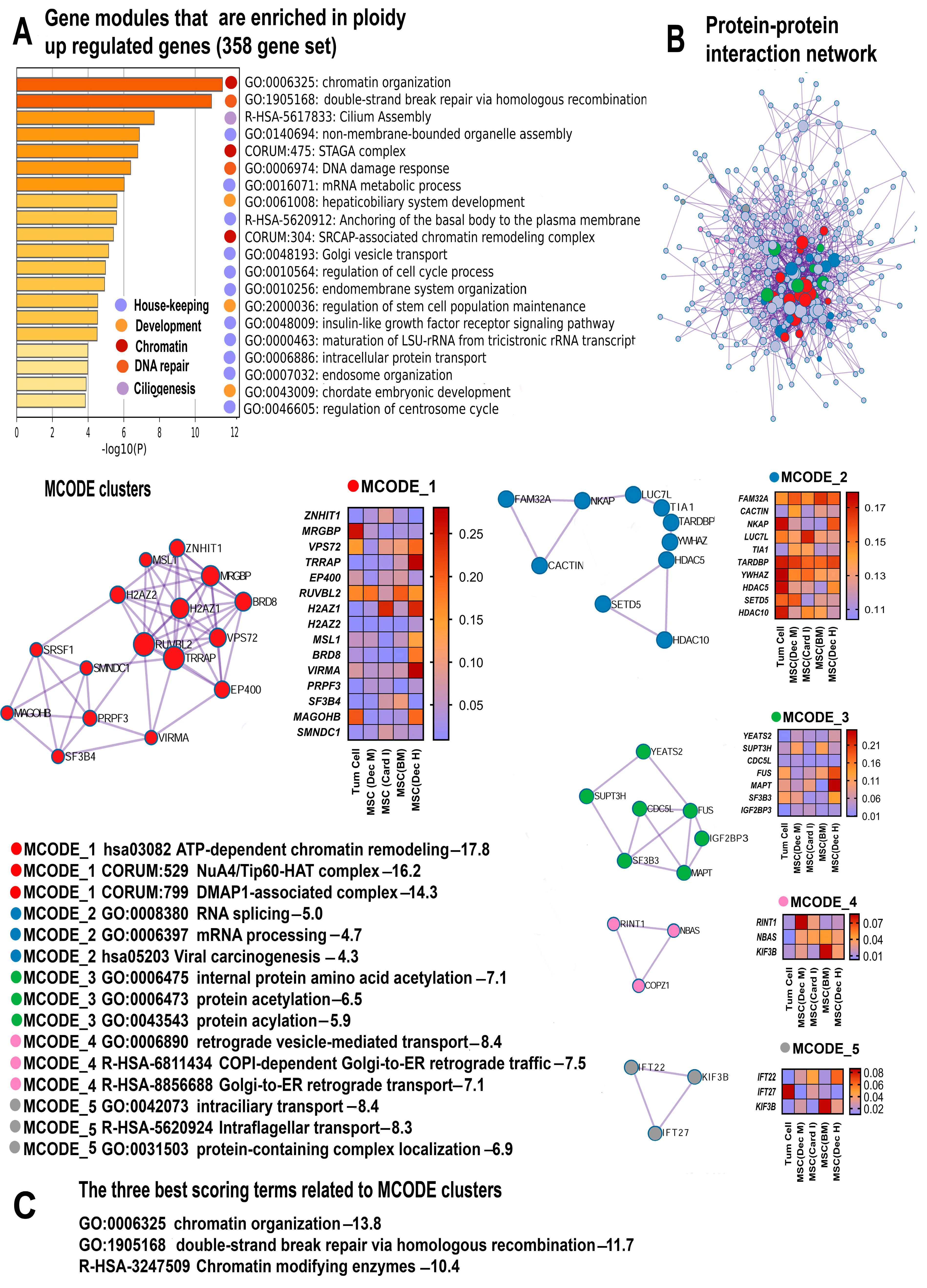
2.2. Gene Modules Upregulated by Polyploidy Reveal Global Transcriptome Activation
2.2.1. Polyploidy Promotes Housekeeping Functions
2.2.2. Polyploidy Reawakens Programs of Unicellularity and Stemness
2.2.3. Polyploidy Promotes Chromatin Opening and Activates Related Double Strand Break DNA Repair Pathways
2.2.4. Polyploidy Boosts Ciliogenesis and Centrosome Cycle
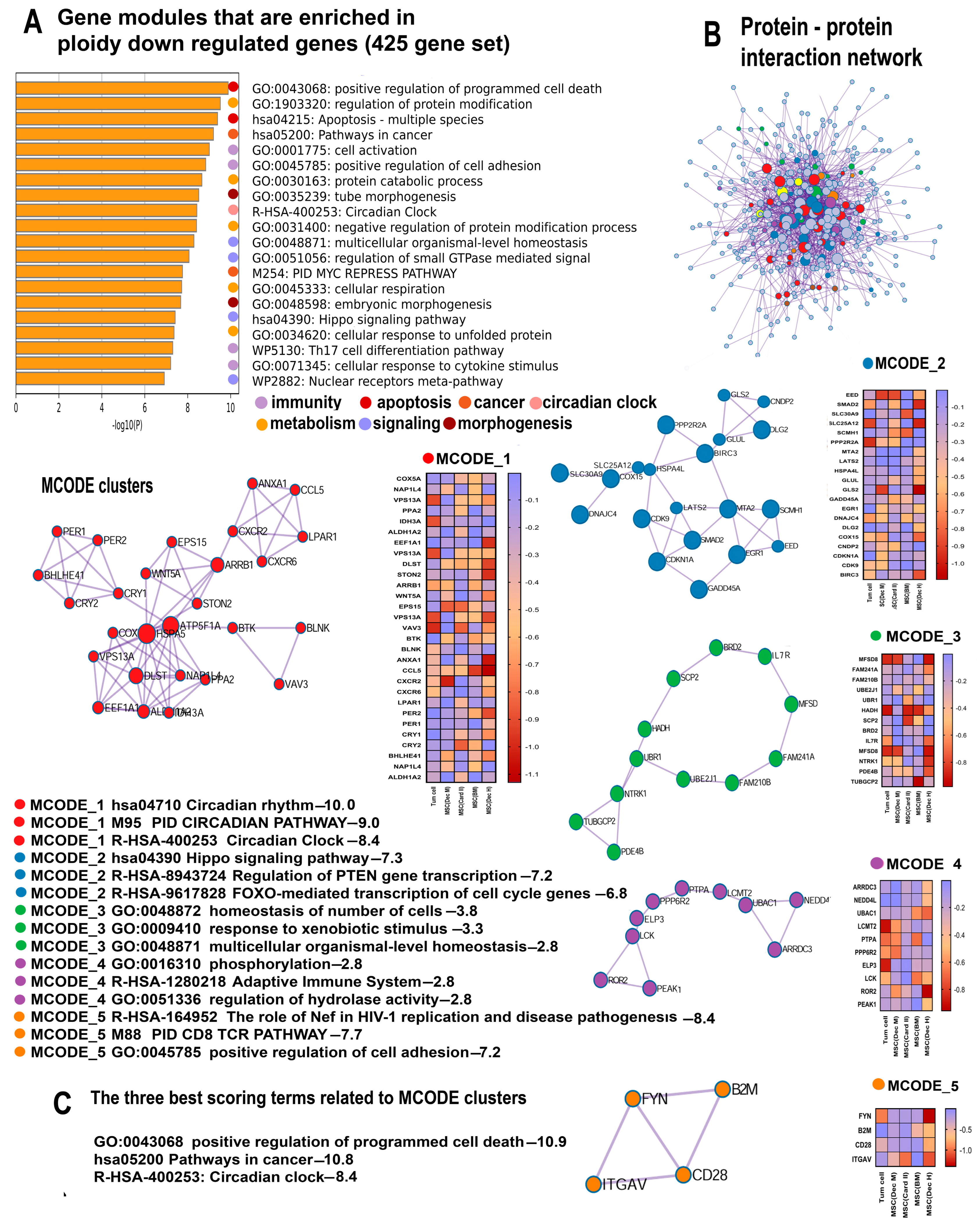
2.3. Polyploidy Downregulates Gene Modules Related to Immunity, Apoptosis, and the Circadian Clock
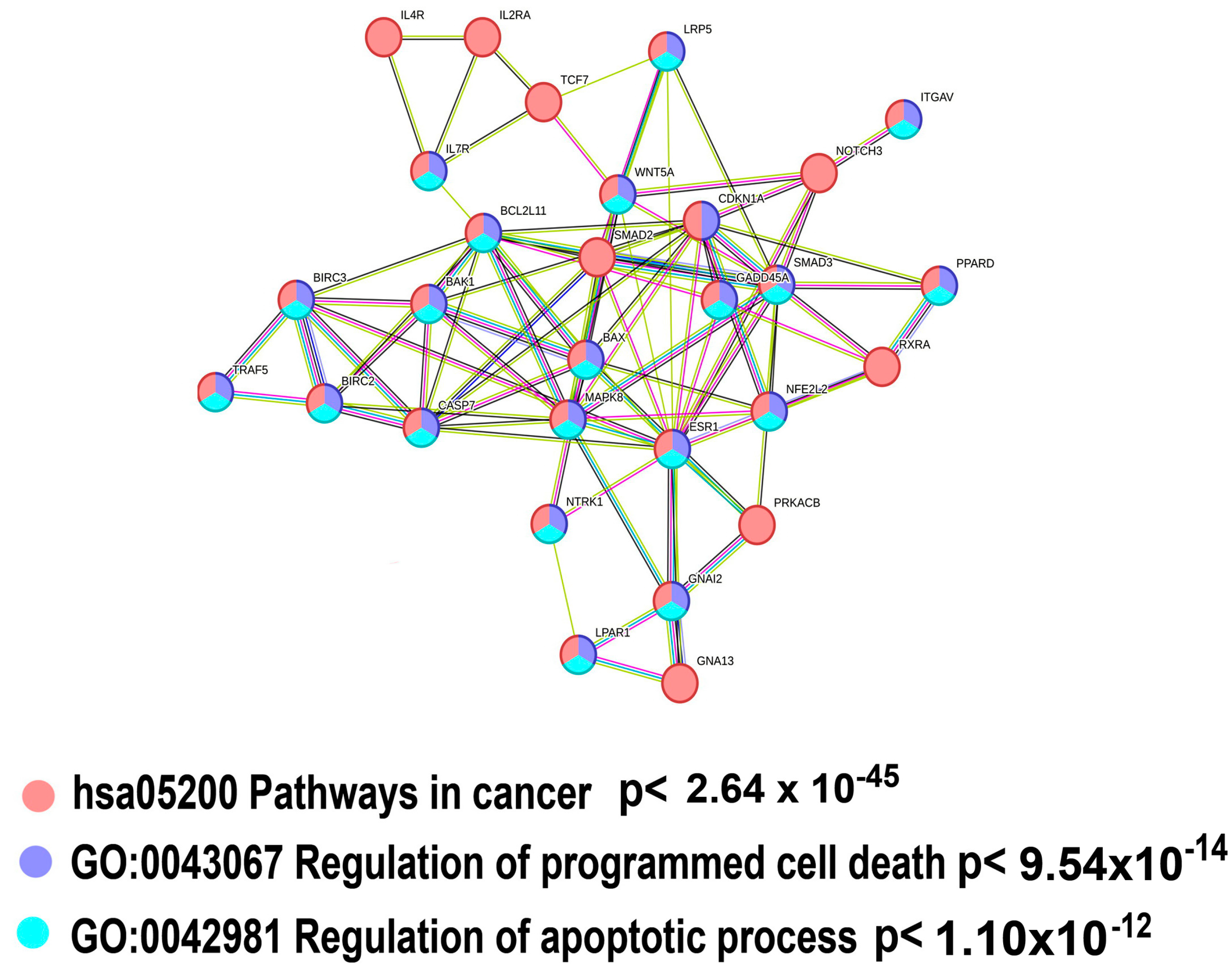
2.3.1. Polyploidy Downregulates Pathways Related to Cell Death and Apoptosis
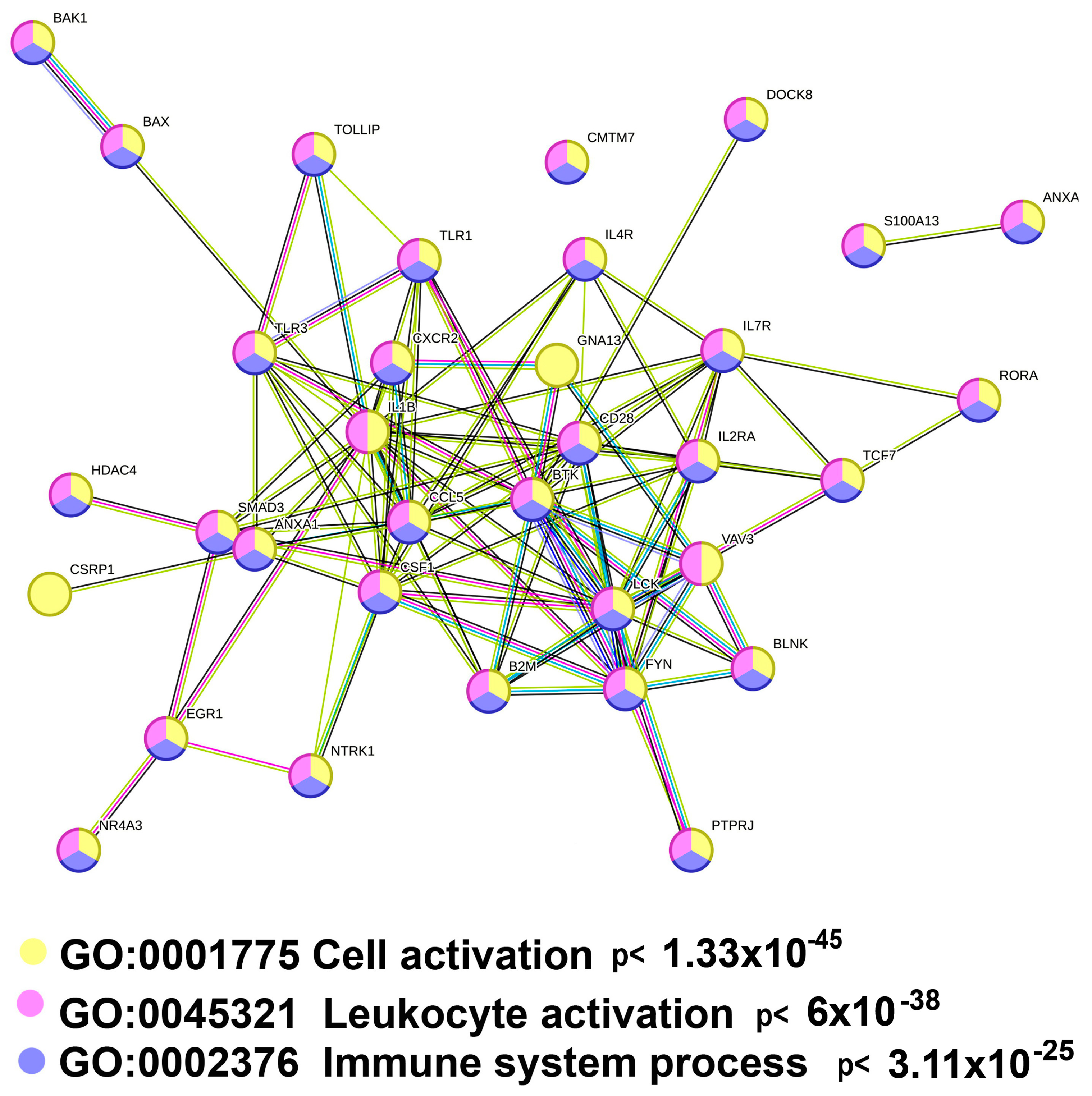
2.3.2. Polyploidy Downregulates the Overlapping Pathways of Cell Activation and Immunity
2.3.3. Polyploidy Attenuates Pathways Related to the Circadian Clock
2.4. The Ploidy-Regulated Genes Derived from Early Cardiac Progenitors and Young Cardiomyocytes Obtained from iPS Demonstrate a Good Agreement with the Results Obtained on Cancer Cells and MSC
3. Discussion
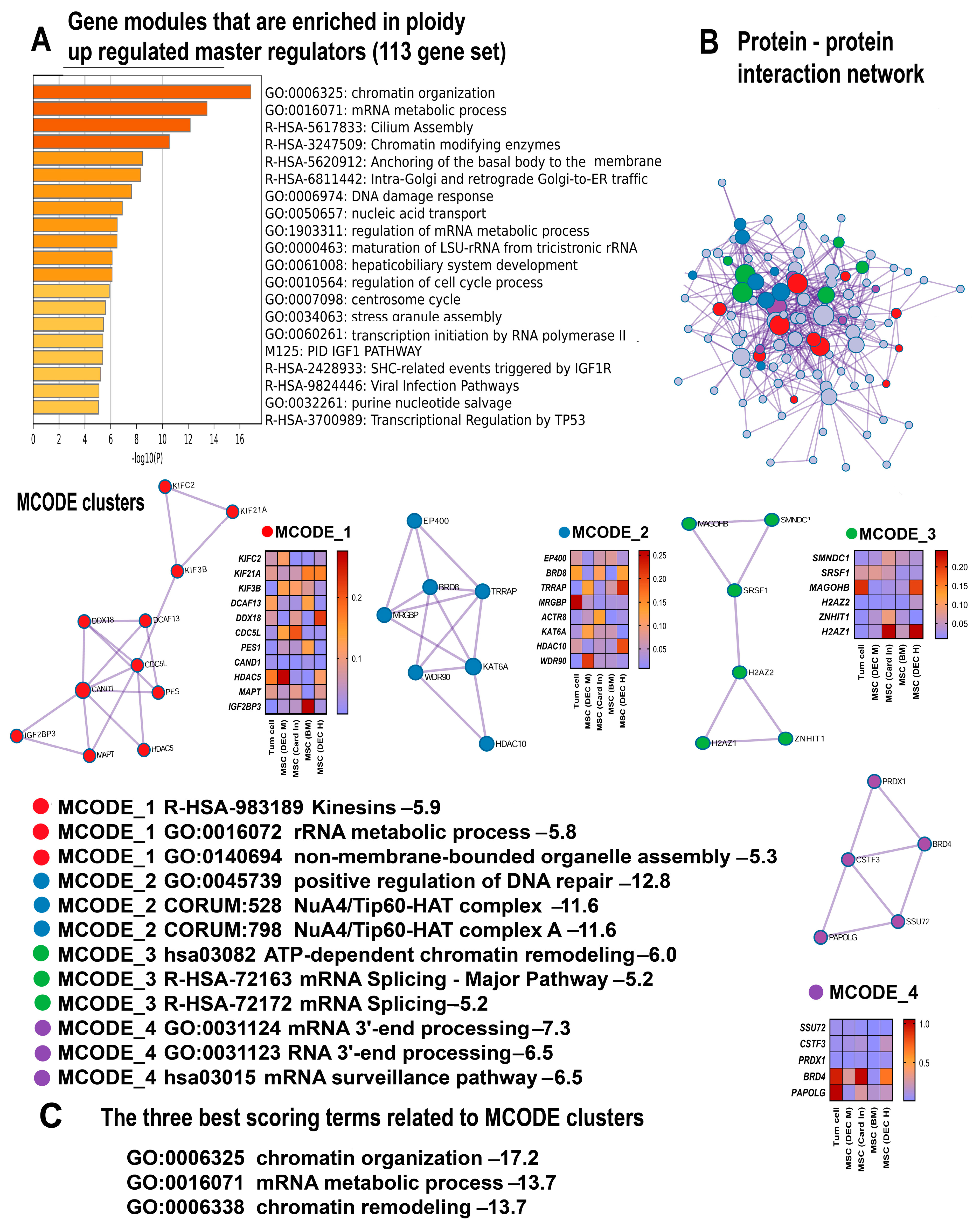
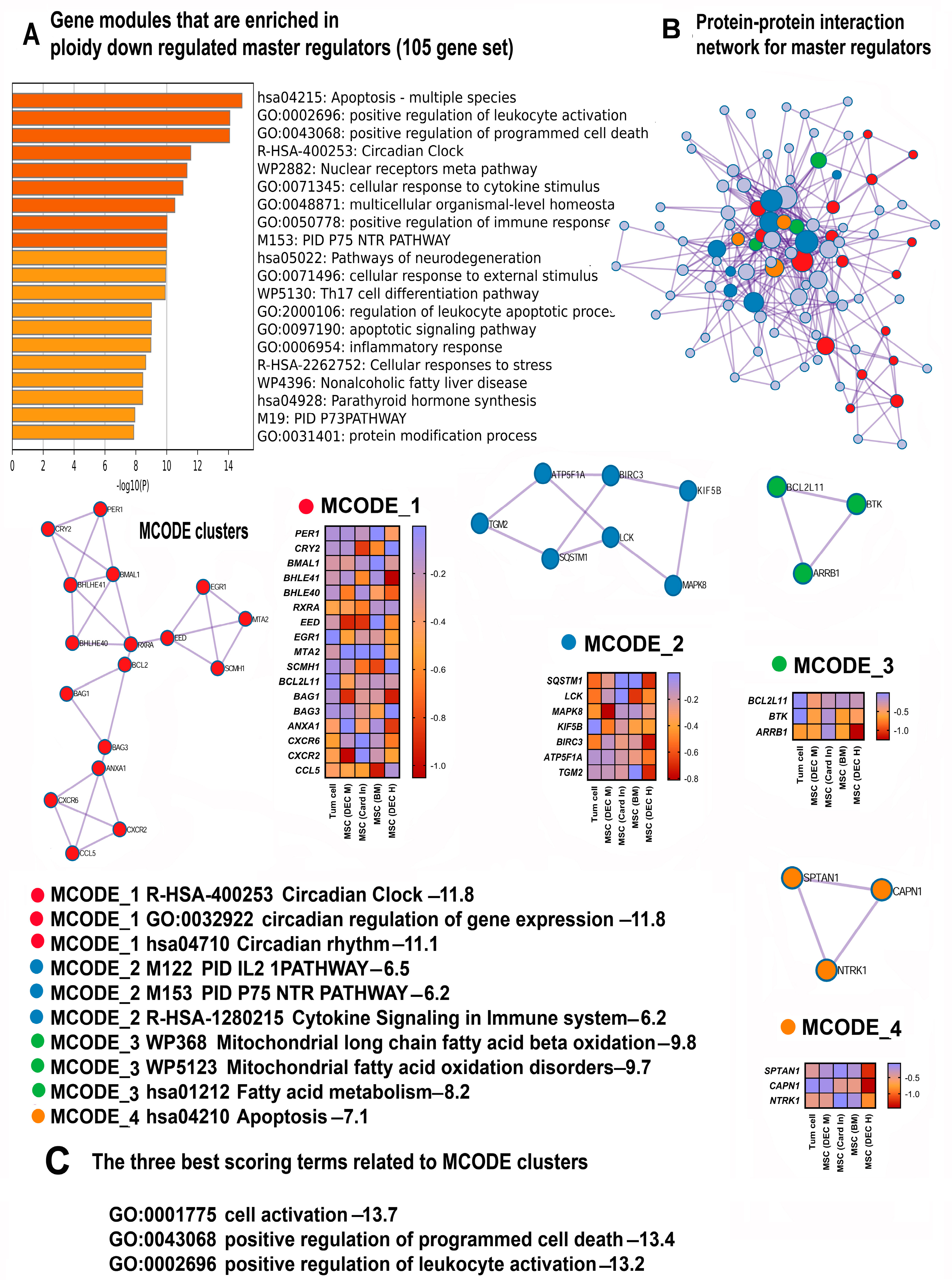
3.1. The Analysis of Consensus Genes and Master Regulators Identified Common Features of Polyploidy across Cancer Cells and Adult Mesenchymal Stem Cells
3.2. Polyploidy Is Associated with a Hypertranscription State
3.3. Polyploidy Promotes Ciliogenesis and the Centrosome Cycle
3.4. Polyploidy Impairs Signaling via the Circadian Clock
4. Materials and Methods
4.1. Databases
4.2. Obtaining the Sets of Ploidy-Induced and Ploidy-Suppressed Consensus Genes
4.3. Obtaining the Sets of Ploidy-Induced or Ploidy-Suppressed Master Regulator Genes
4.4. Enrichment Analysis of All Consensus DEGs and Consensus Master Regulators Associated with Polyploidy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vietri, M.T.; D’Elia, G.; Caliendo, G.; Casamassimi, A.; Federico, A.; Passariello, L.; Cioffi, M.; Molinari, A.M. Prevalence of Mutations in BRCA and MMR Genes in Patients Affected with Hereditary Endometrial Cancer. Med. Oncol. 2021, 38, 13. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Fuentes, D.E.; Fernández-Garza, L.E.; Samia-Meza, J.A.; Barrera-Barrera, S.A.; Caplan, A.I.; Barrera-Saldaña, H.A. Mesenchymal Stem Cells Current Clinical Applications: A Systematic Review. Arch. Med. Res. 2021, 52, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Neri, S. Genetic Stability of Mesenchymal Stromal Cells for Regenerative Medicine Applications: A Fundamental Biosafety Aspect. Int. J. Mol. Sci. 2019, 20, 2406. [Google Scholar] [CrossRef]
- Nimiritsky, P.P.; Eremichev, R.Y.; Alexandrushkina, N.A.; Efimenko, A.Y.; Tkachuk, V.A.; Makarevich, P.I. Unveiling Mesenchymal Stromal Cells’ Organizing Function in Regeneration. Int. J. Mol. Sci. 2019, 20, 823. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal Stem Cell Perspective: Cell Biology to Clinical Progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef]
- Sagaradze, G.D.; Basalova, N.A.; Efimenko, A.Y.; Tkachuk, V.A. Mesenchymal Stromal Cells as Critical Contributors to Tissue Regeneration. Front. Cell Dev. Biol. 2020, 8, 576176. [Google Scholar] [CrossRef] [PubMed]
- Makarevich, P.I.; Hu, Y.-C. Editorial: Regulation of Adult Stem Cells Fate and Function in Natural and Artificial Microenvironments. Front. Cell Dev. Biol. 2022, 10, 955568. [Google Scholar] [CrossRef] [PubMed]
- Makarevich, P.I.; Efimenko, A.Y.; Tkachuk, V.A. Biochemical Regulation of Regenerative Processes by Growth Factors and Cytokines: Basic Mechanisms and Relevance for Regenerative Medicine. Biochemistry 2020, 85, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Kou, M.; Huang, L.; Yang, J.; Chiang, Z.; Chen, S.; Liu, J.; Guo, L.; Zhang, X.; Zhou, X.; Xu, X.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles for Immunomodulation and Regeneration: A next Generation Therapeutic Tool? Cell Death Dis. 2022, 13, 580. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, M.; Nezhad, M.S.; Mehranfar, F.; Golpour, M.; Esakandari, M.A.; Rashmeie, Z.; Ghorbani, M.; Nasimi, F.; Hoseinian, S.N. Biological Aspects and Clinical Applications of Mesenchymal Stem Cells: Key Features You Need to Be Aware of. Curr. Pharm. Biotechnol. 2021, 22, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.; Thangavelu, L.; Aravindhan, S.; Zekiy, A.O.; Jarahian, M.; Chartrand, M.S.; Pathak, Y.; Marofi, F.; Shamlou, S.; Hassanzadeh, A. Mesenchymal Stem/Stromal Cells as a Valuable Source for the Treatment of Immune-Mediated Disorders. Stem Cell Res. Ther. 2021, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem Cells: Past, Present, and Future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, V.; Astrelina, T.; Nugis, V.; Ostashkin, A.; Karaseva, T.; Dobrovolskaya, E.; Usupzhanova, D.; Suchkova, Y.; Lomonosova, E.; Rodin, S.; et al. Clonal Chromosomal and Genomic Instability during Human Multipotent Mesenchymal Stromal Cells Long-Term Culture. PLoS ONE 2018, 13, e0192445. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Ashman, T.-L.; Soltis, P.S.; Soltis, D.E. Polyploidy: An Evolutionary and Ecological Force in Stressful Times. Plant Cell 2021, 33, 11–26. [Google Scholar] [CrossRef] [PubMed]
- De Smet, R.; Van de Peer, Y. Redundancy and Rewiring of Genetic Networks Following Genome-Wide Duplication Events. Curr. Opin. Plant Biol. 2012, 15, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy and Myc Proto-Oncogenes Promote Stress Adaptation via Epigenetic Plasticity and Gene Regulatory Network Rewiring. Int. J. Mol. Sci. 2022, 23, 9691. [Google Scholar] [CrossRef] [PubMed]
- Vittoria, M.A.; Quinton, R.J.; Ganem, N.J. Whole-Genome Doubling in Tissues and Tumors. Trends Genet. 2023, 39, 954–967. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E.; Vainshelbaum, N.M.; Giuliani, A.; Erenpreisa, J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. Int. J. Mol. Sci. 2020, 21, 8759. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Whole-Genome Duplications in Evolution, Ontogeny, and Pathology: Complexity and Emergency Reserves. Mol. Biol. 2021, 55, 927–943. [Google Scholar] [CrossRef]
- Gemble, S.; Wardenaar, R.; Keuper, K.; Srivastava, N.; Nano, M.; Macé, A.-S.; Tijhuis, A.E.; Bernhard, S.V.; Spierings, D.C.J.; Simon, A.; et al. Genetic Instability from a Single S Phase after Whole-Genome Duplication. Nature 2022, 604, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-Genome Doubling Confers Unique Genetic Vulnerabilities on Tumour Cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid Giant Cancer Cells: An Emerging New Field of Cancer Biology. Semin. Cancer Biol. 2022, 81, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Hammarlund, E.U.; Brown, J.S.; Amend, S.R.; Axelrod, R.M. Cancer Recurrence and Lethality Are Enabled by Enhanced Survival and Reversible Cell Cycle Arrest of Polyaneuploid Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2020838118. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Hammarlund, E.U.; Austin, R.H.; Axelrod, R.; Brown, J.S.; Amend, S.R. Cancer Cells Employ an Evolutionarily Conserved Polyploidization Program to Resist Therapy. Semin. Cancer Biol. 2022, 81, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of Cancer: Survival at the Brink. Semin. Cancer Biol. 2022, 81, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Systemic Alterations of Cancer Cells and Their Boost by Polyploidization: Unicellular Attractor (UCA) Model. Int. J. Mol. Sci. 2023, 24, 6196. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Giuliani, A.; Yoshikawa, K.; Falk, M.; Hildenbrand, G.; Salmina, K.; Freivalds, T.; Vainshelbaum, N.; Weidner, J.; Sievers, A.; et al. Spatial-Temporal Genome Regulation in Stress-Response and Cell-Fate Change. Int. J. Mol. Sci. 2023, 24, 2658. [Google Scholar] [CrossRef] [PubMed]
- Fajka-Boja, R.; Marton, A.; Tóth, A.; Blazsó, P.; Tubak, V.; Bálint, B.; Nagy, I.; Hegedűs, Z.; Vizler, C.; Katona, R.L. Increased Insulin-like Growth Factor 1 Production by Polyploid Adipose Stem Cells Promotes Growth of Breast Cancer Cells. BMC Cancer 2018, 18, 872. [Google Scholar] [CrossRef] [PubMed]
- Broughton, K.M.; Khieu, T.; Nguyen, N.; Rosa, M.; Mohsin, S.; Quijada, P.; Wang, B.J.; Echeagaray, O.H.; Kubli, D.A.; Kim, T.; et al. Cardiac Interstitial Tetraploid Cells Can Escape Replicative Senescence in Rodents but Not Large Mammals. Commun. Biol. 2019, 2, 205. [Google Scholar] [CrossRef]
- Fox, D.T.; Soltis, D.E.; Soltis, P.S.; Ashman, T.-L.; Van de Peer, Y. Polyploidy: A Biological Force From Cells to Ecosystems. Trends Cell Biol. 2020, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, O.; Massalha, H.; Shani, N.; Kagan, S.; Ravid, O.; Madar, S.; Trakhtenbrot, L.; Leshkowitz, D.; Rechavi, G.; Zipori, D. Polyploidization of Murine Mesenchymal Cells Is Associated with Suppression of the Long Noncoding RNA H19 and Reduced Tumorigenicity. Cancer Res. 2012, 72, 6403–6413. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Gao, F.; Rusie, A.; Hemingway, J.; Ostmann, A.B.; Sroga, J.M.; Jegga, A.G.; Das, S.K. Decidual Cell Polyploidization Necessitates Mitochondrial Activity. PLoS ONE 2011, 6, e26774. [Google Scholar] [CrossRef] [PubMed]
- Morey, R.; Farah, O.; Kallol, S.; Requena, D.F.; Meads, M.; Moretto-Zita, M.; Soncin, F.; Laurent, L.C.; Parast, M.M. Transcriptomic Drivers of Differentiation, Maturation, and Polyploidy in Human Extravillous Trophoblast. Front. Cell Dev. Biol. 2021, 9, 702046. [Google Scholar] [CrossRef] [PubMed]
- Krane, M.; Dreßen, M.; Santamaria, G.; My, I.; Schneider, C.M.; Dorn, T.; Laue, S.; Mastantuono, E.; Berutti, R.; Rawat, H.; et al. Sequential Defects in Cardiac Lineage Commitment and Maturation Cause Hypoplastic Left Heart Syndrome. Circulation 2021, 144, 1409–1428. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. DNA Helix: The Importance of Being AT-Rich. Mamm. Genome 2017, 28, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Giuliani, A. Networks of Networks: An Essay on Multi-Level Biological Organization. Front. Genet. 2021, 12, 706260. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.; Giuliani, A. Editorial: Prediction and Explanation in Biomedicine Using Network-Based Approaches. Front. Genet. 2022, 13, 967936. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, O.; Helmy, M.; Giuliani, A.; Selvarajoo, K. Globally Invariant Behavior of Oncogenes and Random Genes at Population but Not at Single Cell Level. NPJ Syst. Biol. Appl. 2023, 9, 28. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Bulut-Karslioglu, A.; Ramalho-Santos, M. Hypertranscription in Development, Stem Cells, and Regeneration. Dev. Cell 2017, 40, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Gene Golden Age Paradox and Its Partial Solution. Genomics 2019, 111, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary Framework of the Human Interactome: Unicellular and Multicellular Giant Clusters. Biosystems 2019, 181, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Levanon, E.Y. Human Housekeeping Genes, Revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A Comprehensive Resource for Annotation and Prediction of Animal Transcription Factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered Interactions between Unicellular and Multicellular Genes Drive Hallmarks of Transformation in a Diverse Range of Solid Tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Anatskaya, O.V. Growth of Biological Complexity from Prokaryotes to Hominids Reflected in the Human Genome. Int. J. Mol. Sci. 2021, 22, 11640. [Google Scholar] [CrossRef]
- Medvedeva, Y.A.; Lennartsson, A.; Ehsani, R.; Kulakovskiy, I.V.; Vorontsov, I.E.; Panahandeh, P.; Khimulya, G.; Kasukawa, T.; FANTOM Consortium; Drabløs, F. EpiFactors: A Comprehensive Database of Human Epigenetic Factors and Complexes. Database 2015, 2015, bav067. [Google Scholar] [CrossRef] [PubMed]
- Selenina, A.V.; Tsimokha, A.S.; Tomilin, A.N. Proteasomes in Protein Homeostasis of Pluripotent Stem Cells. Acta Naturae 2017, 9, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, S.A.; Anatskaya, O.V.; Vinogradov, A.E.; Uversky, V.N.; Dayhoff, G.W.; Bystriakova, M.A.; Pospelov, V.A.; Tolkunova, E.N. Isolation and Characterization of Human Colon Adenocarcinoma Stem-Like Cells Based on the Endogenous Expression of the Stem Markers. Int. J. Mol. Sci. 2021, 22, 4682. [Google Scholar] [CrossRef] [PubMed]
- Vainshelbaum, N.M.; Giuliani, A.; Salmina, K.; Pjanova, D.; Erenpreisa, J. The Transcriptome and Proteome Networks of Malignant Tumours Reveal Atavistic Attractors of Polyploidy-Related Asexual Reproduction. Int. J. Mol. Sci. 2022, 23, 14930. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Cancer Genes and Cancer Stem Cells in Tumorigenesis: Evolutionary Deep Homology and Controversies. Genes Dis. 2022, 9, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Kasperski, A. Life Entrapped in a Network of Atavistic Attractors: How to Find a Rescue. Int. J. Mol. Sci. 2022, 23, 4017. [Google Scholar] [CrossRef]
- Skvortsova, E.V.; Sinenko, S.A.; Tomilin, A.N. Immortalized Murine Fibroblast Cell Lines Are Refractory to Reprogramming to Pluripotent State. Oncotarget 2018, 9, 35241–35250. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, E.V.; Nazarov, I.B.; Tomilin, A.N.; Sinenko, S.A. Dual Mode of Mitochondrial ROS Action during Reprogramming to Pluripotency. Int. J. Mol. Sci. 2022, 23, 10924. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Tomilin, A.N. Metabolic Control of Induced Pluripotency. Front. Cell Dev. Biol. 2023, 11, 1328522. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, M.; Escobar, M.; Forero Amézquita, S.; Cubillos, D.; Rincón, C.; Vanegas, P.; Tarazona, M.P.; Atuesta Escobar, S.; Blanco, J.C.; Celis, L.G. Application of the Yamanaka Transcription Factors Oct4, Sox2, Klf4, and c-Myc from the Laboratory to the Clinic. Genes 2023, 14, 1697. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Cho, B.; Cook, D.P.; Trcka, D.; Wrana, J.L.; Ramalho-Santos, M. Absolute Scaling of Single-Cell Transcriptomes Identifies Pervasive Hypertranscription in Adult Stem and Progenitor Cells. Cell Rep. 2023, 42, 111978. [Google Scholar] [CrossRef] [PubMed]
- Tartour, K.; Andriani, F.; Folco, E.G.; Letkova, D.; Schneider, R.; Saidi, I.; Sato, T.; Welz, P.-S.; Benitah, S.A.; Allier, C.; et al. Mammalian PERIOD2 Regulates H2A.Z Incorporation in Chromatin to Orchestrate Circadian Negative Feedback. Nat. Struct. Mol. Biol. 2022, 29, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.; Palhan, V.B.; Tjernberg, A.; Lymar, E.S.; Gamper, A.M.; Kundu, T.K.; Chait, B.T.; Roeder, R.G. Human STAGA Complex Is a Chromatin-Acetylating Transcription Coactivator That Interacts with Pre-mRNA Splicing and DNA Damage-Binding Factors In Vivo. Mol. Cell Biol. 2001, 21, 6782–6795. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Vorontchikhina, M.; Wang, Y.-L.; Faiola, F.; Martinez, E. STAGA Recruits Mediator to the MYC Oncoprotein to Stimulate Transcription and Cell Proliferation. Mol. Cell Biol. 2008, 28, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ichikawa, W.; Faiola, F.; Lo, S.-Y.; Liu, X.; Martinez, E. MYC Interacts with the Human STAGA Coactivator Complex via Multivalent Contacts with the GCN5 and TRRAP Subunits. Biochim. Biophys. Acta 2014, 1839, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Procida, T.; Friedrich, T.; Jack, A.P.M.; Peritore, M.; Bönisch, C.; Eberl, H.C.; Daus, N.; Kletenkov, K.; Nist, A.; Stiewe, T.; et al. JAZF1, A Novel P400/TIP60/NuA4 Complex Member, Regulates H2A.Z Acetylation at Regulatory Regions. Int. J. Mol. Sci. 2021, 22, 678. [Google Scholar] [CrossRef] [PubMed]
- Fréchard, A.; Faux, C.; Hexnerova, R.; Crucifix, C.; Papai, G.; Smirnova, E.; McKeon, C.; Ping, F.L.Y.; Helmlinger, D.; Schultz, P.; et al. The Structure of the NuA4-Tip60 Complex Reveals the Mechanism and Importance of Long-Range Chromatin Modification. Nat. Struct. Mol. Biol. 2023, 30, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Raff, J.W. Centrioles, Centrosomes, and Cilia in Health and Disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic Polyploidy Is Associated with the Upregulation of C-MYC Interacting Genes and EMT-like Signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef] [PubMed]
- Herriage, H.C.; Huang, Y.-T.; Calvi, B.R. The Antagonistic Relationship between Apoptosis and Polyploidy in Development and Cancer. Semin. Cell Dev. Biol. 2024, 156, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Paradoxical Relationship between Protein Content and Nucleolar Activity in Mammalian Cardiomyocytes. Genome 2004, 47, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef] [PubMed]
- Casotti, M.C.; Meira, D.D.; Zetum, A.S.S.; Araújo, B.C.D.; Silva, D.R.C.D.; Santos, E.D.V.W.D.; Garcia, F.M.; Paula, F.D.; Santana, G.M.; Louro, L.S.; et al. Computational Biology Helps Understand How Polyploid Giant Cancer Cells Drive Tumor Success. Genes 2023, 14, 801. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Santoro, R. Regulation and Roles of the Nucleolus in Embryonic Stem Cells: From Ribosome Biogenesis to Genome Organization. Stem Cell Rep. 2020, 15, 1206–1219. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three Steps to the Immortality of Cancer Cells: Senescence, Polyploidy and Self-Renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Giant Cells: Linking McClintock’s Heredity to Early Embryogenesis and Tumor Origin throughout Millennia of Evolution on Earth. Semin. Cancer Biol. 2022, 81, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Kasperski, A.; Kasperska, R. Study on Attractors during Organism Evolution. Sci. Rep. 2021, 11, 9637. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. aCLS Cancers: Genomic and Epigenetic Changes Transform the Cell of Origin of Cancer into a Tumorigenic Pathogen of Unicellular Organization and Lifestyle. Gene 2020, 726, 144174. [Google Scholar] [CrossRef]
- Amend, S.R.; Torga, G.; Lin, K.-C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid Giant Cancer Cells: Unrecognized Actuators of Tumorigenesis, Metastasis, and Resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Giuliani, A.; Zimatore, G.; Erenpreisa, J.; Yoshikawa, K. A Unified Genomic Mechanism of Cell-Fate Change. Results Probl. Cell Differ. 2022, 70, 35–69. [Google Scholar] [CrossRef] [PubMed]
- Jemaà, M.; Daams, R.; Charfi, S.; Mertens, F.; Huber, S.M.; Massoumi, R. Tetraploidization Increases the Motility and Invasiveness of Cancer Cells. Int. J. Mol. Sci. 2023, 24, 13926. [Google Scholar] [CrossRef] [PubMed]
- Hota, S.K.; Bruneau, B.G. ATP-Dependent Chromatin Remodeling during Mammalian Development. Development 2016, 143, 2882–2897. [Google Scholar] [CrossRef] [PubMed]
- Colino-Sanguino, Y.; Clark, S.J.; Valdes-Mora, F. The H2A.Z-Nuclesome Code in Mammals: Emerging Functions. Trends Genet. 2022, 38, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Morrison, O.; Thakur, J. Molecular Complexes at Euchromatin, Heterochromatin and Centromeric Chromatin. Int. J. Mol. Sci. 2021, 22, 6922. [Google Scholar] [CrossRef] [PubMed]
- Ghiraldini, F.G.; Silva, I.S.; Mello, M.L.S. Polyploidy and Chromatin Remodeling in Hepatocytes from Insulin-Dependent Diabetic and Normoglycemic Aged Mice. Cytom. A 2012, 81, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.S.; Ghiraldini, F.G.; Veronezi, G.M.B.; Mello, M.L.S. Polyploidy and Nuclear Phenotype Characteristics of Cardiomyocytes from Diabetic Adult and Normoglycemic Aged Mice. Acta Histochem. 2018, 120, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Lambuta, R.A.; Nanni, L.; Liu, Y.; Diaz-Miyar, J.; Iyer, A.; Tavernari, D.; Katanayeva, N.; Ciriello, G.; Oricchio, E. Whole-Genome Doubling Drives Oncogenic Loss of Chromatin Segregation. Nature 2023, 615, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Lashgari, A.; Kougnassoukou Tchara, P.-E.; Lambert, J.-P.; Côté, J. New Insights into the DNA Repair Pathway Choice with NuA4/TIP60. DNA Repair. 2022, 113, 103315. [Google Scholar] [CrossRef]
- Wang, X.; Lupton, C.; Lauth, A.; Wan, T.C.; Foster, P.; Patterson, M.; Auchampach, J.A.; Lough, J.W. Evidence That the Acetyltransferase Tip60 Induces the DNA Damage Response and Cell-Cycle Arrest in Neonatal Cardiomyocytes. J. Mol. Cell Cardiol. 2021, 155, 88–98. [Google Scholar] [CrossRef]
- Wang, X.; Wan, T.C.; Lauth, A.; Purdy, A.L.; Kulik, K.R.; Patterson, M.; Lough, J.W.; Auchampach, J.A. Conditional Depletion of the Acetyltransferase Tip60 Protects against the Damaging Effects of Myocardial Infarction. J. Mol. Cell Cardiol. 2022, 163, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid Cells Produced after Severe Genotoxic Damage Have the Potential to Repair DNA Double Strand Breaks. J. Cell Sci. 2003, 116, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Bafort, Q.; Mizrachi, E.; Audenaert, P.; Simoens, P.; Van Montagu, M.; Bonte, D.; Van de Peer, Y. The Duplication of Genomes and Genetic Networks and Its Potential for Evolutionary Adaptation and Survival during Environmental Turmoil. Proc. Natl. Acad. Sci. USA 2023, 120, e2307289120. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Giuliani, A. Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. Int. J. Mol. Sci. 2019, 21, 240. [Google Scholar] [CrossRef] [PubMed]
- Derks, W.; Rode, J.; Collin, S.; Rost, F.; Heinke, P.; Hariharan, A.; Pickel, L.; Simonova, I.; Lázár, E.; Graham, E.; et al. A Latent Cardiomyocyte Regeneration Potential in Human Heart Disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Derks, W.; Bergmann, O. Cycling Cardiomyocytes: Scarce but Important in Recovery From Heart Infarction? Circ. Res. 2021, 128, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Engel, F.B. Advances in Heart Regeneration Based on Cardiomyocyte Proliferation and Regenerative Potential of Binucleated Cardiomyocytes and Polyploidization. Clin. Sci. 2019, 133, 1229–1253. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Treatment Resistance of Solid Tumors with a Focus on Polyploid/Senescent Giant Cancer Cells (PGCCs). Int. J. Mol. Sci. 2023, 24, 11534. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Gradistics: An Underappreciated Dimension in Evolutionary Space. Biosystems 2023, 224, 104844. [Google Scholar] [CrossRef] [PubMed]
- Swift, S.K.; Purdy, A.L.; Kolell, M.E.; Andresen, K.G.; Lahue, C.; Buddell, T.; Akins, K.A.; Rau, C.D.; O’Meara, C.C.; Patterson, M. Cardiomyocyte Ploidy Is Dynamic during Postnatal Development and Varies across Genetic Backgrounds. Development 2023, 150, dev201318. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E. Genome Size and Chromatin Condensation in Vertebrates. Chromosoma 2005, 113, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E.; Kudryavtsev, B.N. Cardiomyocyte Ploidy Levels in Birds with Different Growth Rates. J. Exp. Zool. 2001, 289, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Myocyte Ploidy in Heart Chambers of Birds with Different Locomotor Activity. J. Exp. Zool. 2002, 293, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Sidorenko, N.V.; Vinogradov, A.E.; Beyer, T.V. Impact of Neonatal Cryptosporidial Gastroenteritis on Epigenetic Programming of Rat Hepatocytes. Cell Biol. Int. 2007, 31, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Sidorenko, N.V.; Beyer, T.V.; Vinogradov, A.E. Neonatal Cardiomyocyte Ploidy Reveals Critical Windows of Heart Development. Int. J. Cardiol. 2010, 141, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Patterson, M.; Sucov, H.M. Cardiomyocyte Polyploidy and Implications for Heart Regeneration. Annu. Rev. Physiol. 2020, 82, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; De Nicolo, A. The Centrosome and the Primary Cilium: The Yin and Yang of a Hybrid Organelle. Cells 2019, 8, 701. [Google Scholar] [CrossRef]
- Lau, T.Y.; Poon, R.Y.C. Whole-Genome Duplication and Genome Instability in Cancer Cells: Double the Trouble. Int. J. Mol. Sci. 2023, 24, 3733. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Duncan, A.W. The Ploidy State as a Determinant of Hepatocyte Proliferation. Semin. Liver Dis. 2023, 43, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Inoko, A.; Inagaki, M. Cell Cycle Progression by the Repression of Primary Cilia Formation in Proliferating Cells. Cell Mol. Life Sci. 2013, 70, 3893–3905. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Tsiokas, L. Cilia and Cell Cycle Re-Entry: More than a Coincidence. Cell Cycle 2011, 10, 2683–2690. [Google Scholar] [CrossRef] [PubMed]
- Walz, G. Role of Primary Cilia in Non-Dividing and Post-Mitotic Cells. Cell Tissue Res. 2017, 369, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Lake, A.V.R.; China, B.; Szymanska, K.; Wheway, G.; Bell, S.; Morrison, E.; Bond, J.; Johnson, C.A. Racgap1 Knockdown Results in Cells with Multiple Cilia Due to Cytokinesis Failure. Ann. Hum. Genet. 2024, 88, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Cell-Cycle Dependence of Transcriptome Gene Modules: Comparison of Regression Lines. FEBS J. 2020, 287, 4427–4439. [Google Scholar] [CrossRef] [PubMed]
- Nehme, Z.; Pasquereau, S.; Haidar Ahmad, S.; Coaquette, A.; Molimard, C.; Monnien, F.; Algros, M.-P.; Adotevi, O.; Diab Assaf, M.; Feugeas, J.-P.; et al. Polyploid Giant Cancer Cells, Stemness and Epithelial-Mesenchymal Plasticity Elicited by Human Cytomegalovirus. Oncogene 2021, 40, 3030–3046. [Google Scholar] [CrossRef] [PubMed]
- Yanardag, S.; Pugacheva, E.N. Primary Cilium Is Involved in Stem Cell Differentiation and Renewal through the Regulation of Multiple Signaling Pathways. Cells 2021, 10, 1428. [Google Scholar] [CrossRef]
- Kiseleva, A.A.; Nikonova, A.S.; Golemis, E.A. Patterns of Ciliation and Ciliary Signaling in Cancer. Rev. Physiol. Biochem. Pharmacol. 2023, 185, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Moein, S.; Adibi, R.; da Silva Meirelles, L.; Nardi, N.B.; Gheisari, Y. Cancer Regeneration: Polyploid Cells Are the Key Drivers of Tumor Progression. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188408. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Carpenter, V.J.; Bloukh, S.; Gewirtz, D.A. Targeting Tumor Cell Senescence and Polyploidy as Potential Therapeutic Strategies. Semin. Cancer Biol. 2022, 81, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Barriuso, D.; Alvarez-Frutos, L.; Gonzalez-Gutierrez, L.; Motiño, O.; Kroemer, G.; Palacios-Ramirez, R.; Senovilla, L. Involvement of Bcl-2 Family Proteins in Tetraploidization-Related Senescence. Int. J. Mol. Sci. 2023, 24, 6374. [Google Scholar] [CrossRef]
- Choi, B.K.A.; D’Onofrio, P.M.; Shabanzadeh, A.P.; Koeberle, P.D. Stabilization of Primary Cilia Reduces Abortive Cell Cycle Re-Entry to Protect Injured Adult CNS Neurons from Apoptosis. PLoS ONE 2019, 14, e0220056. [Google Scholar] [CrossRef] [PubMed]
- Behnam, B.; Fazilaty, H.; Ghadyani, M.; Fadavi, P.; Taghizadeh-Hesary, F. Ciliated, Mitochondria-Rich Postmitotic Cells Are Immune-Privileged, and Mimic Immunosuppressive Microenvironment of Tumor-Initiating Stem Cells: From Molecular Anatomy to Molecular Pathway. Front. Biosci. (Landmark Ed.) 2023, 28, 261. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H. Primary Cilia: A Novel Research Approach to Overcome Anticancer Drug Resistance. Front. Mol. Biosci. 2023, 10, 1270639. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.M.; Lin, C.-H.; Sun, H.-L.; Alencastro, F.; Zhu, A.C.; Aljuhani, M.; Navari, L.; Yilmaz, S.A.; Yu, L.; Corps, K.; et al. METTL3 Regulates Liver Homeostasis, Hepatocyte Ploidy, and Circadian Rhythm-Controlled Gene Expression in Mice. Am. J. Pathol. 2022, 192, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Vallone, D.; Lahiri, K.; Dickmeis, T.; Foulkes, N.S. Start the Clock! Circadian Rhythms and Development. Dev. Dyn. 2007, 236, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Feillet, C.; van der Horst, G.T.J.; Levi, F.; Rand, D.A.; Delaunay, F. Coupling between the Circadian Clock and Cell Cycle Oscillators: Implication for Healthy Cells and Malignant Growth. Front. Neurol. 2015, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Farshadi, E.; van der Horst, G.T.J.; Chaves, I. Molecular Links between the Circadian Clock and the Cell Cycle. J. Mol. Biol. 2020, 432, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Rosati, D.; Palmieri, M.; Brunelli, G.; Morrione, A.; Iannelli, F.; Frullanti, E.; Giordano, A. Differential Gene Expression Analysis Pipelines and Bioinformatic Tools for the Identification of Specific Biomarkers: A Review. Comput. Struct. Biotechnol. J. 2024, 23, 1154–1168. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W.V. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- An, O.; Pendino, V.; D’Antonio, M.; Ratti, E.; Gentilini, M.; Ciccarelli, F.D. NCG 4.0: The Network of Cancer Genes in the Era of Massive Mutational Screenings of Cancer Genomes. Database 2014, 2014, bau015. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, M.; Wang, D.; Diao, L.; Liu, J.; Tang, L.; Guo, S.; He, F.; Li, D. HisgAtlas 1.0: A Human Immunosuppression Gene Database. Database 2017, 2017, bax094. [Google Scholar] [CrossRef] [PubMed]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A Comprehensive Catalogue of Known and Candidate Cancer Genes from Cancer Sequencing Screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.; Iyer, V.R. Global Identification of Myc Target Genes Reveals Its Direct Role in Mitochondrial Biogenesis and Its E-Box Usage In Vivo. PLoS ONE 2008, 3, e1798. [Google Scholar] [CrossRef] [PubMed]
- Kurum, E.; Benayoun, B.A.; Malhotra, A.; George, J.; Ucar, D. Computational Inference of a Genomic Pluripotency Signature in Human and Mouse Stem Cells. Biol. Direct. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An Updated Literature-Based Knowledgebase for Tumor Suppressor Genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway/Signature | Gene Number | O/E Ratio * | FDR ** |
|---|---|---|---|
| Housekeeping functions | |||
| Organelle biogenesis and maintenance (Reactome) | 16 | 8.18 | 4.78 × 10−08 |
| Housekeeping genes [46] #& | 50 | 2.34 | 2.02 × 10−07 |
| Gene Expression (Reactome) | 33 | 3.17 | 3.64 × 10−07 |
| TF-cofactors Animal TFDB [47] | 23 | 3.88 | 3.58 × 10−06 |
| Intra-Golgi and retrograde Golgi-to-ER traffic (Reactome) | 10 | 9.62 | 1.66 × 10−05 |
| Golgi-to-ER retrograde transport (Reactome) | 7 | 11.00 | 4.43 × 10−04 |
| AURKA Activation by TPX2 (Reactome) | 6 | 14.08 | 5.24 × 10−04 |
| Centrosome maturation; Recruitment of mitotic centrosome proteins and complexes (Reactome) #& | 6 | 12.85 | 7.83 × 10−04 |
| COPI-dependent Golgi-to-ER retrograde traffic (Reactome) # | 6 | 12.69 | 8.16 × 10−04 |
| Regulation of PLK1 Activity at G2/M Transition (Reactome) #& | 6 | 11.68 | 1.21 × 10−03 |
| IGF1 pathway (Pathway Interaction Database) #& | 4 | 24.47 | 1.82 × 10−03 |
| Top 10% AT3 SW-degenerate synonymous sites [37] #& | 27 | 2.33 | 1.88 × 10−03 |
| 3rd PIN cluster [45] #& | 22 | 2.62 | 2.28 × 10−03 |
| Membrane Trafficking (Reactome) | 13 | 3.67 | 4.33 × 10−03 |
| Processing of Capped Intron-Containing Pre-mRNA (Reactome) | 8 | 5.76 | 5.96 × 10−03 |
| mRNA Splicing—Major Pathway (Reactome) | 7 | 6.74 | 6.12 × 10−03 |
| Mitotic G2-G2/M phases (Reactome) | 7 | 6.66 | 6.30 × 10−03 |
| Vesicle-mediated transport (Reactome) | 13 | 3.45 | 6.62 × 10−03 |
| Insulin-like Growth Factor-2 mRNA Binding Proteins (IGF2BPs/IMPs/VICKZs) bind RNA (Reactome) | 2 | 114.18 | 6.62 × 10−03 |
| mRNA Splicing (Reactome) #& | 7 | 6.45 | 7.14 × 10−03 |
| Spliceosome (Kegg) | 6 | 7.73 | 8.24 × 10−03 |
| Top 10% selection-favored in mammals [44] | 21 | 2.38 | 9.03 × 10−03 |
| Cell Cycle, Mitotic (Reactome) | 11 | 3.71 | 1.15 × 10−02 |
| Cell Cycle (Reactome) | 12 | 3.35 | 1.45 × 10−02 |
| Cellular responses to stress (Reactome) | 9 | 3.47 | 5.77 × 10−02 |
| RNA Polymerase II Transcription (Reactome) | 5 | 5.72 | 8.24 × 10−02 |
| Insulin Pathway (Pathway Interaction Database) #& | 3 | 11.95 | 8.60 × 10−02 |
| Unicellularity | |||
| Unicellular genes [48] # | 77 | 2.08 | 1.30 × 10−11 |
| Human-yeast 1:1 orthologs (Ensembl) | 19 | 3.37 | 4.28 × 10−04 |
| Unicellular genes [49] #& | 52 | 1.75 | 6.14 × 10−04 |
| Unicellular PIN cluster [44] | 36 | 1.94 | 3.26 × 10−03 |
| Myc signaling and stemness | |||
| MYC interactants (String) | 15 | 3.18 | 5.61 × 10−03 |
| Kit receptor signaling pathway (Reactome) | 4 | 2.18 | 5.48 × 10−03 |
| HALLMARK_MYC_TARGETS (Molecular Signatures Database) # | 7 | 5.99 | 1.06 × 10−02 |
| C-MYC pathway (Pathway Interaction Database) #& | 3 | 23.35 | 1.57 × 10−02 |
| Chromatin and DNA damage response | |||
| EpiFactors database [50] #& | 26 | 6.23 | 2.73 × 10−11 |
| Chromatin organization; Chromatin modifying enzymes (Reactome) #& | 14 | 8.72 | 2.41 × 10−07 |
| HATs acetylate histones (Reactome) #& | 8 | 9.65 | 2.87 × 10−04 |
| HALLMARK_DNA_REPAIR (Molecular Signatures Database) # | 8 | 9.13 | 3.96 × 10−04 |
| DNA Damage/Telomere Stress Induced Senescence (Reactome) # | 4 | 8.56 | 5.95 × 10−02 |
| Epigenetic regulation of gene expression (Reactome) # | 5 | 5.79 | 8.05 × 10−02 |
| Ciliogenesis | |||
| Cilium Assembly (Reactome) #& | 14 | 12.75 | 2.39 × 10−09 |
| Anchoring of the basal body to the plasma membrane (Reactome) # | 9 | 15.73 | 1.43 × 10−06 |
| Loss of proteins required for interphase microtubule organization from the centrosome; Loss of Nlp from mitotic centrosomes (Reactome) # | 6 | 14.68 | 4.41 × 10−04 |
| Pathway/Signature | Gene Number | O/E * Ratio | FDR ** |
|---|---|---|---|
| Immunity | |||
| HALLMARK_TNFA_SIGNALING_VIA_NFKB (Molecular Signatures Database) | 18 | 16.11 | 2.03 × 10−14 |
| Immune System (Reactome) | 39 | 3.36 | 7.21 × 10−10 |
| HALLMARK_INFLAMMATORY_RESPONSE (Molecular Signatures Database) | 14 | 12.53 | 1.39 × 10−09 |
| Regulation of toll-like receptor signaling pathway (WikiPathways) | 11 | 14.17 | 5.53 × 10−08 |
| Cytokine Signaling in Immune system (Reactome) | 21 | 5.07 | 1.12 × 10−07 |
| Innate Immune System (Reactome) | 26 | 3.67 | 6.75 × 10−07 |
| Toll-like Receptor Signaling Pathway (WikiPathways) | 8 | 14.04 | 1.07 × 10−05 |
| Toll-like receptor signaling pathway (Kegg) | 8 | 13.77 | 1.21 × 10−05 |
| Signaling by Interleukins (Reactome) | 15 | 5.21 | 1.62 × 10−05 |
| Thymic Stromal LymphoPoietin (TSLP) Signaling Pathway (WikiPathways) | 6 | 22.86 | 2.01 × 10−05 |
| IL2-mediated signaling events (Pathway Interaction Database) | 6 | 20.66 | 3.22 × 10−05 |
| HALLMARK_IL2_STAT5_SIGNALING (Molecular Signatures Database) | 9 | 8.10 | 1.11 × 10−04 |
| Toll-Like Receptors Cascades (Reactome) | 8 | 9.55 | 1.17 × 10−04 |
| TNF related weak inducer of apoptosis (TWEAK) Signaling Pathway (WikiPathways) | 5 | 21.32 | 2.07 × 10−04 |
| DAP12 signaling (Reactome) | 11 | 5.60 | 2.54 × 10−04 |
| DAP12 interactions (Reactome) | 11 | 5.37 | 3.65 × 10−04 |
| HALLMARK_ALLOGRAFT_REJECTION (Molecular Signatures Database) | 10 | 8.95 | 1.62 × 10−05 |
| TNF signaling pathway (Kegg) | 6 | 9.95 | 1.41 × 10−03 |
| Fc epsilon receptor (FCERI) signaling (Reactome) | 10 | 4.87 | 1.67 × 10−03 |
| Activated TLR4 signaling (Reactome) | 6 | 9.51 | 1.67 × 10−03 |
| Apoptosis and cell death | |||
| Apoptosis—multiple species (Kegg) | 9 | 50.36 | 2.96 × 10−11 |
| Apoptosis Modulation and Signaling (WikiPathways) | 10 | 19.68 | 1.54 × 10−08 |
| Apoptosis (Kegg) | 10 | 12.98 | 6.75 × 10−07 |
| Programmed Cell Death (Reactome) | 10 | 10.47 | 4.72 × 10−06 |
| Intrinsic Pathway for Apoptosis (Reactome) | 6 | 24.98 | 1.33 × 10−05 |
| HALLMARK_APOPTOSIS (Molecular Signatures Database) | 9 | 10.01 | 2.48 × 10−05 |
| Apoptosis (Reactome) | 9 | 9.59 | 3.22 × 10−05 |
| Apoptosis (WikiPathways) | 7 | 14.92 | 3.22 × 10−05 |
| Apoptosis-related network due to altered Notch3 in ovarian cancer (WikiPathways) | 6 | 20.27 | 3.48 × 10−05 |
| Caspase cascade in apoptosis (Pathway Interaction Database) | 5 | 17.91 | 4.58 × 10−04 |
| Circadian clock | 9 | 35.03 | 8.55 × 10−10 |
| BMAL1:CLOCK, NPAS2 activates circadian gene expression (Reactome) | 9 | 35.03 | 8.55 × 10−10 |
| Circadian Clock (Reactome) | 9 | 23.35 | 2.77 × 10−08 |
| Circadian rhythm related genes (WikiPathways) | 12 | 10.69 | 1.91 × 10−07 |
| Circadian rhythm (Kegg) | 5 | 28.88 | 5.12 × 10−05 |
| Circadian rhythm related genes (WikiPathways) | 12 | 10.69 | 1.91 × 10−07 |
| Tumor suppressor genes | 23 | 4.22 | 4.85 × 10−07 |
| TSGene downregulated pancancer (Tumor Suppressor Gene database | 23 | 4.22 | 4.85 × 10−07 |
| TSGene all (Tumor Suppressor Gene database) | 23 | 4.06 | 9.07 × 10−07 |
| Validated targets of C-MYC transcriptional repression (Kegg) | 5 | 11.74 | 8.97 × 10−04 |
| Hippo signaling pathway (Kegg) | 12 | 5.45 | 6.05 × 10−03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anatskaya, O.V.; Vinogradov, A.E. Polyploidy Promotes Hypertranscription, Apoptosis Resistance, and Ciliogenesis in Cancer Cells and Mesenchymal Stem Cells of Various Origins: Comparative Transcriptome In Silico Study. Int. J. Mol. Sci. 2024, 25, 4185. https://doi.org/10.3390/ijms25084185
Anatskaya OV, Vinogradov AE. Polyploidy Promotes Hypertranscription, Apoptosis Resistance, and Ciliogenesis in Cancer Cells and Mesenchymal Stem Cells of Various Origins: Comparative Transcriptome In Silico Study. International Journal of Molecular Sciences. 2024; 25(8):4185. https://doi.org/10.3390/ijms25084185
Chicago/Turabian StyleAnatskaya, Olga V., and Alexander E. Vinogradov. 2024. "Polyploidy Promotes Hypertranscription, Apoptosis Resistance, and Ciliogenesis in Cancer Cells and Mesenchymal Stem Cells of Various Origins: Comparative Transcriptome In Silico Study" International Journal of Molecular Sciences 25, no. 8: 4185. https://doi.org/10.3390/ijms25084185
APA StyleAnatskaya, O. V., & Vinogradov, A. E. (2024). Polyploidy Promotes Hypertranscription, Apoptosis Resistance, and Ciliogenesis in Cancer Cells and Mesenchymal Stem Cells of Various Origins: Comparative Transcriptome In Silico Study. International Journal of Molecular Sciences, 25(8), 4185. https://doi.org/10.3390/ijms25084185