
Experimental Models to Study Immune Dysfunction in the Pathogenesis of Parkinson’s Disease

 , ,
, ,  ,
,  , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
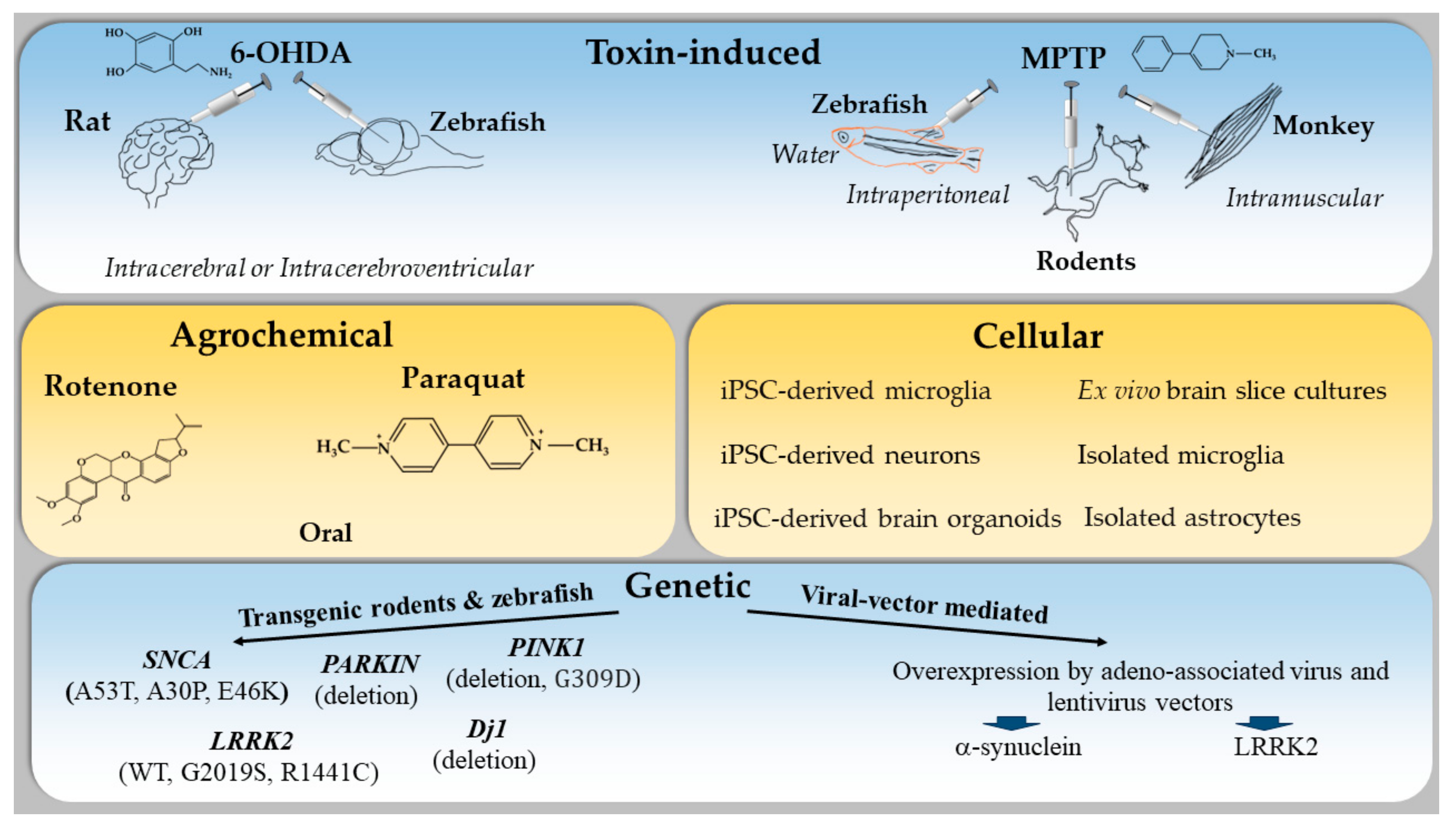
2. Animal Models to Study the Immune System in PD
2.1. Toxin-Induced Models of PD
2.1.1. 6-Hydroxydopamine-Induced (6-OHDA) Model of PD
2.1.2. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-Induced Model of PD
2.1.3. Advantages and Disadvantages of Toxin-Induced Experimental PD
2.2. Genetic Models of PD
2.3. Agrochemical-Induced Models of PD
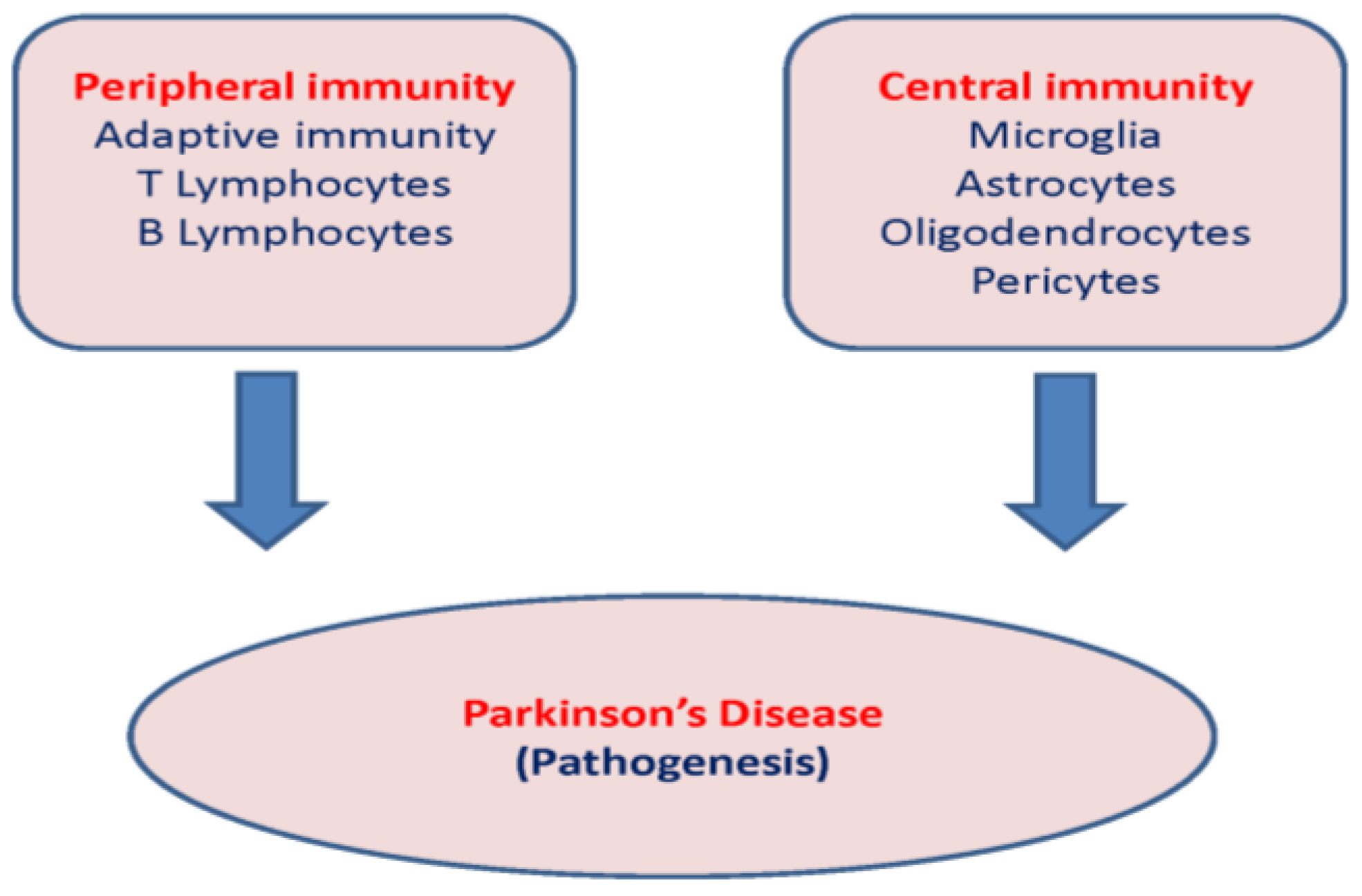
3. Peripheral Immunity in the Pathogenesis of PD
3.1. Adaptive Immunity
3.1.1. T Lymphocytes in PD
3.1.2. Antibodies and B Lymphocytes in PD
4. Central Immunity
4.1. PD and Resident Immune Cells
4.2. Tissue Infiltration
4.3. Model to Assess Tissue Infiltration
5. Other Models to Study Immune Dysfunction in PD
5.1. Zebrafish
5.2. Cellular Models
5.2.1. Microglial Cell Cultures
5.2.2. Culture of Peripheral Immune Cells: Monocytes–Lymphocytes
5.2.3. Co-Culture Models
5.2.4. Ex Vivo Brain Slice Cultures with Immune Cells
5.2.5. Organoids
6. BBB Alterations in PD and the Immune Response
7. Glymphatic System and PD
8. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s Disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Rocca, W.A. Time Trends in the Incidence of Parkinson Disease. JAMA Neurol. 2016, 73, 981. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Terkelsen, M.H.; Klaestrup, I.H.; Hvingelby, V.; Lauritsen, J.; Pavese, N.; Romero-Ramos, M. Neuroinflammation and Immune Changes in Prodromal Parkinson’s Disease and Other Synucleinopathies. J. Park. Dis. 2022, 12, S149–S163. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Golombek, D. Circadian and Sleep Disorders in Parkinson’s Disease. Exp. Neurol. 2013, 243, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-Motor Features of Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-First versus Body-First Parkinson’s Disease: A Multimodal Imaging Case-Control Study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P. The Brain-First vs. Body-First Model of Parkinson’s Disease with Comparison to Alternative Models. J. Neural Transm. 2023, 130, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Berg, D.; Gasser, T.; Chen, H.; Yao, C.; Postuma, R.B. Update of the MDS Research Criteria for Prodromal Parkinson’s Disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Ikuno, M.; Yamakado, H.; Takahashi, R. Animal Model for Prodromal Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1961. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Nehru, B. Inhibition of Neuroinflammation and Mitochondrial Dysfunctions by Carbenoxolone in the Rotenone Model of Parkinson’s Disease. Mol. Neurobiol. 2015, 51, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.M.G.; Becegato, M.; Meurer, Y.S.R.; Lima, A.C.; Gonçalves, N.; Bioni, V.S.; Engi, S.A.; Bianchi, P.C.; Cruz, F.C.; Santos, J.R.; et al. Neuroinflammation in Early, Late and Recovery Stages in a Progressive Parkinsonism Model in Rats. Front. Neurosci. 2022, 16. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, U. 6-Hydroxy-Dopamine Induced Degeneration of Central Monoamine Neurons. Eur. J. Pharmacol. 1968, 5, 107–110. [Google Scholar] [CrossRef]
- Ciric, J.; Kapor, S.; Perovic, M.; Saponjic, J. Alterations of Sleep and Sleep Oscillations in the Hemiparkinsonian Rat. Front. Neurosci. 2019, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Zeljkovic Jovanovic, M.; Stanojevic, J.; Stevanovic, I.; Stekic, A.; Bolland, S.J.; Jasnic, N.; Ninkovic, M.; Zaric Kontic, M.; Ilic, T.V.; Rodger, J.; et al. Intermittent Theta Burst Stimulation Improves Motor and Behavioral Dysfunction through Modulation of NMDA Receptor Subunit Composition in Experimental Model of Parkinson’s Disease. Cells 2023, 12, 1525. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Przedborski, S. Parkinson’s Disease: Animal Models and Dopaminergic Cell Vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal Models of Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Porter, C.C.; Totaro, J.A.; Stone, C.A. Effect of 6-Hydroxydopamine and Some Other Compounds on the Concentration of Norepinephrine in the Hearts of Mice. J. Pharmacol. Exp. Ther. 1963, 140, 308–316. [Google Scholar] [PubMed]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [PubMed]
- Chotibut, T.; Apple, D.M.; Jefferis, R.; Salvatore, M.F. Dopamine Transporter Loss in 6-OHDA Parkinson’s Model Is Unmet by Parallel Reduction in Dopamine Uptake. PLoS ONE 2012, 7, e52322. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kumar, S.; Dikshit, M. Involvement of the Mitochondrial Apoptotic Pathway and Nitric Oxide Synthase in Dopaminergic Neuronal Death Induced by 6-Hydroxydopamine and Lipopolysaccharide. Redox Rep. 2010, 15, 115. [Google Scholar] [CrossRef] [PubMed]
- Glinka, Y.; Gassen, M.; Youdim, M.B.H. Mechanism of 6-Hydroxydopamine Neurotoxicity. J. Neural Transm. Suppl. 1997, 50, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Díaz, M.; Abdala, P.; Barroso-Chinea, P.; Obeso, J.; González-Hernández, T. Motor Behavioural Changes after Intracerebroventricular Injection of 6-Hydroxydopamine in the Rat: An Animal Model of Parkinson’s Disease. Behav. Brain Res. 2001, 122, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.S.; Jackson-Lewis, V.; Burke, R.E. 6-Hydroxydopamine Lesion of the Rat Substantia Nigra: Time Course and Morphology of Cell Death. Neurodegeneration 1995, 4, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Rosenblad, C.; Björklund, A. Characterization of Behavioral and Neurodegenerative Changes Following Partial Lesions of the Nigrostriatal Dopamine System Induced by Intrastriatal 6-Hydroxydopamine in the Rat. Exp. Neurol. 1998, 152, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Evenden, J.L.; Robbins, T.W. Depletion of Unilateral Striatal Dopamine Impairs Initiation of Contralateral Actions and Not Sensory Attention. Nature 1985, 313, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Cousins, M.S.; Sokolowski, J.D.; Salamone, J.D. Different Effects of Nucleus Accumbens and Ventrolateral Striatal Dopamine Depletions on Instrumental Response Selection in the Rat. Pharmacol. Biochem. Behav. 1993, 46, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Dunnett, S.B.; Iversen, S.D. Sensorimotor Impairments Following Localized Kainic Acid and 6-Hydroxydopamine Lesions of the Neostriatum. Brain Res. 1982, 248, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, R.E.; Hess, A.; Duvoisin, R.C. Dopaminergic Neurotoxicity of 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyridine in Mice. Science 1984, 224, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-Linked Oxidation in Brain Mitochondria by 1-Methyl-4-Phenyl-Pyridine, a Metabolite of the Neurotoxin, 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef] [PubMed]
- Markey, S.P.; Johannessen, J.N.; Chiueh, C.C.; Burns, R.S.; Herkenham, M.A. Intraneuronal Generation of a Pyridinium Metabolite May Cause Drug-Induced Parkinsonism. Nature 1984, 311, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Riachi, N.J.; LaManna, J.C.; Harik, S.I. Entry of 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine into the Rat Brain. J. Pharmacol. Exp. Ther. 1989, 249, 744–748. [Google Scholar]
- Bezard, E.; Gross, C.E.; Fournier, M.C.; Dovero, S.; Bloch, B.; Jaber, M. Absence of MPTP-Induced Neuronal Death in Mice Lacking the Dopamine Transporter. Exp. Neurol. 1999, 155, 268–273. [Google Scholar] [CrossRef]
- Davey, G.P.; Tipton, K.F.; Murphy, M.P. Uptake and Accumulation of 1-Methyl-4-Phenylpyridinium by Rat Liver Mitochondria Measured Using an Ion-Selective Electrode. Biochem. J. 1992, 288, 439. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; St Clair, D.; Wermer, M.; Yen, H.C.; Oberley, T.; Yang, L.; Flint Beal, M. Manganese Superoxide Dismutase Overexpression Attenuates MPTP Toxicity. Neurobiol. Dis. 1998, 5, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Tieu, K.; Guégan, C.; Caspersen, C.; Jackson-Lewis, V.; Carelli, V.; Martinuzzi, A.; Hirano, M.; Przedborski, S.; Vila, M. Complex I Deficiency Primes Bax-Dependent Neuronal Apoptosis through Mitochondrial Oxidative Damage. Proc. Natl. Acad. Sci. USA 2005, 102, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Bové, J.; Vila, M. Mitochondria and Programmed Cell Death in Parkinson’s Disease: Apoptosis and Beyond. Antioxid. Redox Signal. 2012, 16, 883–895. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M. The 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model. Ann. N. Y. Acad. Sci. 2003, 991, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Giovanni, A.; Sonsalla, P.K.; Heikkila, R.E. Studies on Species Sensitivity to the Dopaminergic Neurotoxin 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine. Part 2: Central Administration of 1-Methyl-4-Phenylpyridinium. J. Pharmacol. Exp. Ther. 1994, 270, 1008–1014. [Google Scholar] [PubMed]
- Prediger, R.D.S.; Rial, D.; Medeiros, R.; Figueiredo, C.P.; Doty, R.L.; Takahashi, R.N. Risk Is in the Air: An Intranasal MPTP (1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Rat Model of Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2009, 1170, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Jakowec, M.; Burke, R.E.; Przedborski, S. Time Course and Morphology of Dopaminergic Neuronal Death Caused by the Neurotoxin 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine. Neurodegeneration 1995, 4, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Naini, A.B.; Jakowec, M.; Petzinger, G.; Miller, R.; Akram, M. The Parkinsonian Toxin 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP): A Technical Review of Its Utility and Safety. J. Neurochem. 2001, 76, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Tatton, N.A.; Kish, S.J. In Situ Detection of Apoptotic Nuclei in the Substantia Nigra Compacta of 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Treated Mice Using Terminal Deoxynucleotidyl Transferase Labelling and Acridine Orange Staining. Neuroscience 1997, 77, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Wahlsten, D.; Metten, P.; Phillips, T.J.; Boehm, S.L.; Burkhart-Kasch, S.; Dorow, J.; Doerksen, S.; Downing, C.; Fogarty, J.; Rodd-Henricks, K.; et al. Different Data from Different Labs: Lessons from Studies of Gene-Environment Interaction. J. Neurobiol. 2003, 54, 283–311. [Google Scholar] [CrossRef] [PubMed]
- Krackow, S.; Vannoni, E.; Codita, A.; Mohammed, A.H.; Cirulli, F.; Branchi, I.; Alleva, E.; Reichelt, A.; Willuweit, A.; Voikar, V.; et al. Consistent Behavioral Phenotype Differences between Inbred Mouse Strains in the IntelliCage. Genes Brain Behav. 2010, 9, 722–731. [Google Scholar] [CrossRef]
- Jiang, P.E.; Lang, Q.H.; Yu, Q.Y.; Tang, X.Y.; Liu, Q.Q.; Li, X.Y.; Feng, X.Z. Behavioral Assessments of Spontaneous Locomotion in a Murine MPTP-Induced Parkinson’s Disease Model. J. Vis. Exp. 2019, 2019, e58653. [Google Scholar] [CrossRef]
- Sedelis, M.; Schwarting, R.K.W.; Huston, J.P. Behavioral Phenotyping of the MPTP Mouse Model of Parkinson’s Disease. Behav. Brain Res. 2001, 125, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Willis, G.L.; Donnan, G.A. Histochemical, Biochemical and Behavioural Consequences of MPTP Treatment in C-57 Black Mice. Brain Res. 1987, 402, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wang, Z.; Tong, J.; Wang, M.; Wang, J.; Xu, J.; Bai, X.; Li, H.; Huang, Y.; Wu, Y.; et al. Long-Term Changes in the Nigrostriatal Pathway in the MPTP Mouse Model of Parkinson’s Disease. Neuroscience 2018, 369, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, J.; Radovanovic, L.; Saponjic, J. Prodromal Local Sleep Disorders in a Rat Model of Parkinson’s Disease Cholinopathy, Hemiparkinsonism and Hemiparkinsonism with Cholinopathy. Behav. Brain Res. 2021, 397, 112957. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.; Coulson, E.J.; Rajnarayanan, R.; Oster, H.; Videnovic, A.; Rawashdeh, O. Sleep and Circadian Rhythms in Parkinson’s Disease and Preclinical Models. Mol. Neurodegener. 2022, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Tronci, E.; Francardo, V. Animal Models of L-DOPA-Induced Dyskinesia: The 6-OHDA-Lesioned Rat and Mouse. J. Neural Transm. 2018, 125, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Del-Bel, E.; Padovan-Neto, F.E.; Szawka, R.E.; da-Silva, C.A.; Raisman-Vozari, R.; Anselmo-Franci, J.; Romano-Dutra, A.C.; Guimaraes, F.S. Counteraction by Nitric Oxide Synthase Inhibitor of Neurochemical Alterations of Dopaminergic System in 6-OHDA-Lesioned Rats under L-DOPA Treatment. Neurotox. Res. 2014, 25, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Kuter, K.; Olech, Ł.; Głowacka, U. Prolonged Dysfunction of Astrocytes and Activation of Microglia Accelerate Degeneration of Dopaminergic Neurons in the Rat Substantia Nigra and Block Compensation of Early Motor Dysfunction Induced by 6-OHDA. Mol. Neurobiol. 2018, 55, 3049–3066. [Google Scholar] [CrossRef] [PubMed]
- Oliynyk, Z.; Rudyk, M.; Dovbynchuk, T.; Dzubenko, N.; Tolstanova, G.; Skivka, L. Inflammatory Hallmarks in 6-OHDA- and LPS-Induced Parkinson’s Disease in Rats. Brain Behav. Immun. Health 2023, 30, 100616. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Baltazar, D.; Zavala-Flores, L.M.; Villanueva-Olivo, A. The 6-Hydroxydopamine Model and Parkinsonian Pathophysiology: Novel Findings in an Older Model. Neurologia 2017, 32, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Ramonet, D.; Perier, C. Mitochondrial Alterations in Parkinson’s Disease: New Clues. J. Neurochem. 2008, 107, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Shin, W.-H.; Baek, J.Y.; Cho, E.J.; Baik, H.H.; Kim, S.R.; Won, S.-Y.; Jin, B.K. CB2 Receptor Activation Prevents Glial-Derived Neurotoxic Mediator Production, BBB Leakage and Peripheral Immune Cell Infiltration and Rescues Dopamine Neurons in the MPTP Model of Parkinson’s Disease. Exp. Mol. Med. 2016, 48, e205. [Google Scholar] [CrossRef] [PubMed]
- Parillaud, V.R.; Lornet, G.; Monnet, Y.; Privat, A.L.; Haddad, A.T.; Brochard, V.; Bekaert, A.; de Chanville, C.B.; Hirsch, E.C.; Combadière, C.; et al. Analysis of Monocyte Infiltration in MPTP Mice Reveals That Microglial CX3CR1 Protects against Neurotoxic Over-Induction of Monocyte-Attracting CCL2 by Astrocytes. J. Neuroinflammation 2017, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.L.; Santoro, M.; Mustafa, S.; Riedel, G.; Forrester, J.V.; Teismann, P. Evidence for a Role of Adaptive Immune Response in the Disease Pathogenesis of the MPTP Mouse Model of Parkinson’s Disease. Glia 2016, 64, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Kurkowska-Jastrzebska, I.; Wrońska, A.; Kohutnicka, M.S.; Czlonkowski, A.; Czlonkowska, A. The Inflammatory Reaction Following 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Intoxication in Mouse. Exp. Neurol. 1999, 156, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Date, I.; Felten, D.L.; Felten, S.Y. Long-Term Effect of MPTP in the Mouse Brain in Relation to Aging: Neurochemical and Immunocytochemical Analysis. Brain Res. 1990, 519, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Rothblat, D.S.; Schroeder, J.A.; Schneider, J.S. Tyrosine Hydroxylase and Dopamine Transporter Expression in Residual Dopaminergic Neurons: Potential Contributors to Spontaneous Recovery from Experimental Parkinsonism. J. Neurosci. Res. 2001, 65, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Aznavour, N.; Cendres-Bozzi, C.; Lemoine, L.; Buda, C.; Sastre, J.-P.; Mincheva, Z.; Zimmer, L.; Lin, J.-S. MPTP Animal Model of Parkinsonism: Dopamine Cell Death or Only Tyrosine Hydroxylase Impairment? A Study Using PET Imaging, Autoradiography, and Immunohistochemistry in the Cat. CNS Neurosci. Ther. 2012, 18, 934–941. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, J.P.; Sriram, K.; Miller, D.B. Defining “Neuroinflammation”. Ann. N. Y. Acad. Sci. 2008, 1139, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Machado, V.; Zöller, T.; Attaai, A.; Spittau, B. Microglia-Mediated Neuroinflammation and Neurotrophic Factor-Induced Protection in the MPTP Mouse Model of Parkinson’s Disease-Lessons from Transgenic Mice. Int. J. Mol. Sci. 2016, 17, 151. [Google Scholar] [CrossRef] [PubMed]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Lama, J.; Buhidma, Y.; Fletcher, E.J.R.; Duty, S. Animal Models of Parkinson’s Disease: A Guide to Selecting the Optimal Model for Your Research. Neuronal Signal. 2021, 5, NS20210026. [Google Scholar] [CrossRef] [PubMed]
- Volta, M.; Melrose, H. LRRK2 Mouse Models: Dissecting the Behavior, Striatal Neurochemistry and Neurophysiology of PD Pathogenesis. Biochem. Soc. Trans. 2017, 45, 113–122. [Google Scholar] [CrossRef]
- Aniszewska, A.; Bergström, J.; Ingelsson, M.; Ekmark-Lewén, S. Modeling Parkinson’s Disease-Related Symptoms in Alpha-Synuclein Overexpressing Mice. Brain Behav. 2022, 12, e2628. [Google Scholar] [CrossRef]
- El-Gamal, M.; Salama, M.; Collins-Praino, L.E.; Baetu, I.; Fathalla, A.M.; Soliman, A.M.; Mohamed, W.; Moustafa, A.A. Neurotoxin-Induced Rodent Models of Parkinson’s Disease: Benefits and Drawbacks. Neurotox. Res. 2021, 39, 897–923. [Google Scholar] [CrossRef]
- Schonhoff, A.M.; Williams, G.P.; Wallen, Z.D.; Standaert, D.G.; Harms, A.S. Innate and Adaptive Immune Responses in Parkinson’s Disease. Prog. Brain Res. 2020, 252, 169–216. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Swat, W.; Alt, F.W. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell 2002, 109, S45–S55. [Google Scholar] [CrossRef] [PubMed]
- Vitetta, E.S.; Berton, M.T.; Burger, C.; Kepron, M.; Lee, W.T.; Yin, X.M. Memory B and T Cells. Annu. Rev. Immunol. 1991, 9, 193–217. [Google Scholar] [CrossRef]
- Miman, O.; Kusbeci, O.Y.; Aktepe, O.C.; Cetinkaya, Z. The Probable Relation between Toxoplasma Gondii and Parkinson’s Disease. Neurosci. Lett. 2010, 475, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, J.; Hoogendoorn, H.; Tarnopolsky, M.; Muñoz, D.G. Monoclonal Antibodies against Epstein-Barr Virus Cross-React with Alpha-Synuclein in Human Brain. Neurology 2000, 55, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Ravenholt, R.T.; Foege, W.H. 1918 Influenza, Encephalitis Lethargica, Parkinsonism. Lancet 1982, 2, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.; McClaren, J.; Pani, A.K.; Smeyne, M.; Korff, A.; Webster, R.; Smeyne, R.J. Inflammatory Effects of Highly Pathogenic H5N1 Influenza Virus Infection in the CNS of Mice. J. Neurosci. 2012, 32, 1545–1559. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Watanabe, M.; Itoh, S.; Kuwahara, H.; Hattori, F. Japanese Encephalitis and Parkinsonism. J. Neurol. 1993, 240, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Leta, V.; Urso, D.; Batzu, L.; Lau, Y.H.; Mathew, D.; Boura, I.; Raeder, V.; Falup-Pecurariu, C.; van Wamelen, D.; Ray Chaudhuri, K. Viruses, Parkinsonism and Parkinson’s Disease: The Past, Present and Future. J. Neural Transm. 2022, 129, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, K.M.; Wang, C. Post-Infectious Neurological Disorders. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420952901. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Paulus, K.; Arru, G.; Piredda, R.; Sechi, G.P.; Sechi, L.A. Humoral Cross Reactivity between α-Synuclein and Herpes Simplex-1 Epitope in Parkinson’s Disease, a Triggering Role in the Disease? J. Neuroimmunol. 2016, 291, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Paulus, K.; Galleri, G.; Arru, G.; Manetti, R.; Sechi, G.P.; Sechi, L.A. Homologous HSV1 and Alpha-Synuclein Peptides Stimulate a T Cell Response in Parkinson’s Disease. J. Neuroimmunol. 2017, 310, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Ene, L. Human Immunodeficiency Virus in the Brain-Culprit or Facilitator? Infect. Dis. 2018, 11, 117863371775268. [Google Scholar] [CrossRef] [PubMed]
- Budka, H. Human Immunodeficiency Virus (HIV) Envelope and Core Proteins in CNS Tissues of Patients with the Acquired Immune Deficiency Syndrome (AIDS). Acta Neuropathol. 1990, 79, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Brach-Werner, R. Astrocytes: HIV Cellular Reservoirs and Important Participants in Neuropathogenesis. AIDS 1999, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, E.D.A.; Achim, C.L.; Ayyavoo, V. Immunodetection of Human Immunodeficiency Virus Type 1 (HIV-1) Vpr in Brain Tissue of HIV-1 Encephalitic Patients. J. Neurovirol. 2006, 12, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Cantó-Nogués, C.; Sánchez-Ramón, S.; Álvarez, S.; Lacruz, C.; Muñóz-Fernández, M.Á. HIV-1 Infection of Neurons Might Account for Progressive HIV-1-Associated Encephalopathy in Children. J. Mol. Neurosci. 2005, 27, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Van der Most, R.G.; Murali-Krishna, K.; Ahmed, R. Prolonged Presence of Effector-Memory CD8 T Cells in the Central Nervous System after Dengue Virus Encephalitis. Int. Immunol. 2003, 15, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Cose, S.; Brammer, C.; Khanna, K.M.; Masopust, D.; Lefrançois, L. Evidence That a Significant Number of Naive T Cells Enter Non-Lymphoid Organs as Part of a Normal Migratory Pathway. Eur. J. Immunol. 2006, 36, 1423–1433. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Lira, A.; Kulczycki, J.; Slack, R.; Anisman, H.; Park, D.S. Involvement of the Fc Gamma Receptor in a Chronic N-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Dopaminergic Loss. J. Biol. Chem. 2011, 286, 28783–28793. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted Overexpression of Human Alpha-Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Kannarkat, G.T.; Boss, J.M.; Tansey, M.G. The Role of Innate and Adaptive Immunity in Parkinson’s Disease. J. Park. Dis. 2013, 3, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, M.J.; Weisel, F. Germinal Center Selection and the Development of Memory B and Plasma Cells. Immunol. Rev. 2012, 247, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Nagele, E.; DeMarshall, C.; Acharya, N.; Nagele, R. Diagnosis of Parkinson’s Disease Based on Disease-Specific Autoantibody Profiles in Human Sera. PLoS ONE 2012, 7, e32383. [Google Scholar] [CrossRef] [PubMed]
- Besong-Agbo, D.; Wolf, E.; Jessen, F.; Oechsner, M.; Hametner, E.; Poewe, W.; Reindl, M.; Oertel, W.H.; Noelker, C.; Bacher, M.; et al. Naturally Occurring α-Synuclein Autoantibody Levels Are Lower in Patients with Parkinson Disease. Neurology 2013, 80, 169–175. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; McGeer, E.G. Expression of the Histocompatibility Glycoprotein HLA-DR in Neurological Disease. Acta Neuropathol. 1988, 76, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Le, W.D.; Xie, W.J.; Alexianu, M.E.; Engelhardt, J.I.; Siklós, L.; Appel, S.H. Experimental Destruction of Substantia Nigra Initiated by Parkinson Disease Immunoglobulins. Arch. Neurol. 1998, 55, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Le, W.D.; Appel, S.H. Role of Fcγ Receptors in Nigral Cell Injury Induced by Parkinson Disease Immunoglobulin Injection into Mouse Substantia Nigra. Exp. Neurol. 2002, 176, 322–327. [Google Scholar] [CrossRef]
- Bae, E.J.; Lee, H.J.; Rockenstein, E.; Ho, D.H.; Park, E.B.; Yang, N.Y.; Desplats, P.; Masliah, E.; Lee, S.J. Antibody-Aided Clearance of Extracellular α-Synuclein Prevents Cell-to-Cell Aggregate Transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, Z.; Xu, S.; Liu, Q.; Liu, C.; Luo, Y.; Jin, L.; Li, S. Immune Profiling of Parkinson’s Disease Revealed Its Association With a Subset of Infiltrating Cells and Signature Genes. Front. Aging Neurosci. 2021, 13, 605970. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; I Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Chen, Z.; Trapp, B.D. Microglia and Neuroprotection. J. Neurochem. 2016, 136, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Brück, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and Alpha-Synuclein in Neurodegeneration: A Complex Interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef]
- Gustot, A.; Gallea, J.I.; Sarroukh, R.; Celej, M.S.; Ruysschaert, J.M.; Raussens, V. Amyloid Fibrils Are the Molecular Trigger of Inflammation in Parkinson’s Disease. Biochem. J. 2015, 471, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; McNamara, N.B.; Gerrits, E.; Marzin, M.C.; Kooistra, S.M.; Miron, V.E.; Nutma, E. White Matter Microglia Heterogeneity in the CNS. Acta Neuropathol. 2022, 143, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Chen, S.; Zhu, J.Y.; Ma, J.; Zhai, Y. The Emerging Role of Central and Peripheral Immune Systems in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 872134. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does Neuroinflammation Fan the Flame in Neurodegenerative Diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Tansey, M.G. A Survey from 2012 of Evidence for the Role of Neuroinflammation in Neurotoxin Animal Models of Parkinson’s Disease and Potential Molecular Targets. Exp. Neurol. 2014, 256, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.T.; Kim, K.S.; Kim, E.S.; Lee, S.; Kim, J.; Hoe, H.S.; Kim, D.G. Emerging Pathogenic Role of Peripheral Blood Factors Following BBB Disruption in Neurodegenerative Disease. Ageing Res. Rev. 2021, 68, 101333. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-Synuclein Induces Toll-like Receptor 4 Dependent Inflammatory Responses in Astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 Astrocyte Conversion by Microglia Is Neuroprotective in Models of Parkinson’s Disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Boccazzi, M.; Van Steenwinckel, J.; Schang, A.L.; Faivre, V.; Le Charpentier, T.; Bokobza, C.; Csaba, Z.; Verderio, C.; Fumagalli, M.; Mani, S.; et al. The Immune-Inflammatory Response of Oligodendrocytes in a Murine Model of Preterm White Matter Injury: The Role of TLR3 Activation. Cell Death Dis. 2021, 12, 166. [Google Scholar] [CrossRef] [PubMed]
- Zeis, T.; Enz, L.; Schaeren-Wiemers, N. The Immunomodulatory Oligodendrocyte. Brain Res. 2016, 1641, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Boccazzi, M.; Fumagalli, M. Oligodendrocyte Dysfunction in Amyotrophic Lateral Sclerosis: Mechanisms and Therapeutic Perspectives. Cells 2021, 10, 565. [Google Scholar] [CrossRef]
- Castellani, G.; Schwartz, M. Immunological Features of Non-Neuronal Brain Cells: Implications for Alzheimer’s Disease Immunotherapy. Trends Immunol. 2020, 41, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, Á.; Molnár, K.; Nógrádi, B.; Hernádi, Z.; Nyúl-Tóth, Á.; Wilhelm, I.; Krizbai, I.A. Neurovascular Inflammaging in Health and Disease. Cells 2020, 9, 1614. [Google Scholar] [CrossRef]
- Vedam-Mai, V. Harnessing the Immune System for the Treatment of Parkinson’s Disease. Brain Res. 2021, 1758, 147308. [Google Scholar] [CrossRef] [PubMed]
- Procter, T.V.; Williams, A.; Montagne, A. Interplay between Brain Pericytes and Endothelial Cells in Dementia. Am. J. Pathol. 2021, 191, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.T.; Maki, T.; Ihara, M.; Lee, V.M.Y.; Trojanowski, J.Q. Brain Microvascular Pericytes in Vascular Cognitive Impairment and Dementia. Front. Aging Neurosci. 2020, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Gu, R.; Zhang, M.; Ren, H.; Shu, Q.; Xu, G.; Wu, H. Microglia Enhanced the Angiogenesis, Migration and Proliferation of Co-Cultured RMECs. BMC Ophthalmol. 2018, 18, 249. [Google Scholar] [CrossRef]
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of Pattern Recognition Receptors and Activation of the Non-Canonical Inflammasome Pathway in Brain Pericytes. Brain Behav. Immun. 2017, 64, 220–231. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Montagne, A.; Kisler, K.; Dai, Z.; Wang, Y.; Huuskonen, M.T.; Sagare, A.P.; Lazic, D.; Sweeney, M.D.; Kong, P.; et al. Pericyte Loss Leads to Circulatory Failure and Pleiotrophin Depletion Causing Neuron Loss. Nat. Neurosci. 2019, 22, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric α-Synuclein Induces Blood-Brain Barrier Dysfunction through Activated Brain Pericytes Releasing Inflammatory Mediators In Vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.W.; Klaus, L.C.; Karikari, A.A.; Visanji, N.P.; Brotchie, J.M.; Lang, A.E.; Volkmann, J.; Koprich, J.B. AAV1/2-Induced Overexpression of A53T-α-Synuclein in the Substantia Nigra Results in Degeneration of the Nigrostriatal System with Lewy-like Pathology and Motor Impairment: A New Mouse Model for Parkinson’s Disease. Acta Neuropathol. Commun. 2017, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Karikari, A.A.; McFleder, R.L.; Ribechini, E.; Blum, R.; Bruttel, V.; Knorr, S.; Gehmeyr, M.; Volkmann, J.; Brotchie, J.M.; Ahsan, F.; et al. Neurodegeneration by α-Synuclein-Specific T Cells in AAV-A53T-α-Synuclein Parkinson’s Disease Mice. Brain Behav. Immun. 2022, 101, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Meuwissen, R.; Genc, S.; Genc, K. Inflammation in Parkinson’s Disease. Adv. Protein Chem. Struct. Biol. 2012, 88, 69–132. [Google Scholar] [CrossRef] [PubMed]
- Lucot, K.L.; Stevens, M.Y.; Bonham, T.A.; Azevedo, E.C.; Chaney, A.M.; Webber, E.D.; Jain, P.; Klockow, J.L.; Jackson, I.M.; Carlson, M.L.; et al. Tracking Innate Immune Activation in a Mouse Model of Parkinson’s Disease Using TREM1 and TSPO PET Tracers. J. Nucl. Med. 2022, 63, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, A.M.; Figge, D.A.; Williams, G.P.; Jurkuvenaite, A.; Gallups, N.J.; Childers, G.M.; Webster, J.M.; Standaert, D.G.; Goldman, J.E.; Harms, A.S. Border-Associated Macrophages Mediate the Neuroinflammatory Response in an Alpha-Synuclein Model of Parkinson Disease. Nat. Commun. 2023, 14, 3754. [Google Scholar] [CrossRef] [PubMed]
- Marinova-Mutafchieva, L.; Sadeghian, M.; Broom, L.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Relationship between Microglial Activation and Dopaminergic Neuronal Loss in the Substantia Nigra: A Time Course Study in a 6-Hydroxydopamine Model of Parkinson’s Disease. J. Neurochem. 2009, 110, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-Hydroxydopamine Model of Parkinson’s Disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Schwab, C.; Parent, A.; Doudet, D. Presence of Reactive Microglia in Monkey Substantia Nigra Years after 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Administration. Ann. Neurol. 2003, 54, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Cicchetti, F.; Lapointe, N.; Roberge-Tremblay, A.; Saint-Pierre, M.; Jimenez, L.; Ficke, B.W.; Gross, R.E. Systemic Exposure to Paraquat and Maneb Models Early Parkinson’s Disease in Young Adult Rats. Neurobiol. Dis. 2005, 20, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.; La Vecchia, C.; Nicotera, P. Paraquat and Parkinson’s Disease. Cell Death Differ. 2010, 17, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Chakrabarti, N.; Bhattacharyya, A. Differential Regional Expression Patterns of α-Synuclein, TNF-α, and IL-1β and Variable Status of Dopaminergic Neurotoxicity in Mouse Brain after Paraquat Treatment. J. Neuroinflammation 2011, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Gupta, S.P.; Srivastava, G.; Srivastava, P.K.; Singh, M.P. Role of Secondary Mediators in Caffeine-Mediated Neuroprotection in Maneb- and Paraquat-Induced Parkinson’s Disease Phenotype in the Mouse. Neurochem. Res. 2012, 37, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Gutiérrez, M.T.; Serrano-García, N.; Orozco-Ibarra, M. Rotenone-Induced Model of Parkinson’s Disease: Beyond Mitochondrial Complex I Inhibition. Mol. Neurobiol. 2023, 60, 1929–1948. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective Microglial Activation in the Rat Rotenone Model of Parkinson’s Disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Yin, D.; Zhao, H.; Zhang, L. The Immunology of Parkinson’s Disease. Semin. Immunopathol. 2022, 44, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Depboylu, C.; Stricker, S.; Ghobril, J.P.; Oertel, W.H.; Priller, J.; Höglinger, G.U. Brain-Resident Microglia Predominate over Infiltrating Myeloid Cells in Activation, Phagocytosis and Interaction with T-Lymphocytes in the MPTP Mouse Model of Parkinson Disease. Exp. Neurol. 2012, 238, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Samantaray, S.; Knaryan, V.H.; Shields, D.C.; Cox, A.A.; Haque, A.; Banik, N.L. Inhibition of Calpain Activation Protects MPTP-Induced Nigral and Spinal Cord Neurodegeneration, Reduces Inflammation, and Improves Gait Dynamics in Mice. Mol. Neurobiol. 2015, 52, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Matsui, A.; Ohyagi, M.; Kikutake, C.; Harada, Y.; Iizuka-Koga, M.; Suyama, M.; Yoshimura, A.; Ito, M. In Vitro Generation of Brain Regulatory T Cells by Co-Culturing with Astrocytes. Front. Immunol. 2022, 13, 960036. [Google Scholar] [CrossRef] [PubMed]
- Kozina, E.; Sadasivan, S.; Jiao, Y.; Dou, Y.; Ma, Z.; Tan, H.; Kodali, K.; Shaw, T.; Peng, J.; Smeyne, R.J. Mutant LRRK2 Mediates Peripheral and Central Immune Responses Leading to Neurodegeneration In Vivo. Brain 2018, 141, 1753–1769. [Google Scholar] [CrossRef] [PubMed]
- Tentillier, N.; Etzerodt, A.; Olesen, M.N.; Rizalar, F.S.; Jacobsen, J.; Bender, D.; Moestrup, S.K.; Romero-Ramos, M. Anti-Inflammatory Modulation of Microglia via CD163-Targeted Glucocorticoids Protects Dopaminergic Neurons in the 6-OHDA Parkinson’s Disease Model. J. Neurosci. 2016, 36, 9375–9390. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Doudet, D.D.; Schwab, C.; Yu, S.; McGeer, E.G.; McGeer, P.L. Role of ICAM-1 in Persisting Inflammation in Parkinson Disease and MPTP Monkeys. Exp. Neurol. 2006, 197, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Bakay, R.A.E.; Boyer, K.L.; Freed, C.R.; Ansari, A.A. Immunological Responses to Injury and Grafting in the Central Nervous System of Nonhuman Primates. Cell Transplant. 1998, 7, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Zwi, S.F.; Choron, C.; Zheng, D.; Nguyen, D.; Zhang, Y.; Roshal, C.; Kikuchi, K.; Hesselson, D. Pharmacological Enhancement of Regeneration-Dependent Regulatory T Cell Recruitment in Zebrafish. Int. J. Mol. Sci. 2019, 20, 5189. [Google Scholar] [CrossRef] [PubMed]
- Konstantin Nissen, S.; Farmen, K.; Carstensen, M.; Schulte, C.; Goldeck, D.; Brockmann, K.; Romero-Ramos, M. Changes in CD163+, CD11b+, and CCR2+ Peripheral Monocytes Relate to Parkinson’s Disease and Cognition. Brain Behav. Immun. 2022, 101, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Mazzolini, J.; Le Clerc, S.; Morisse, G.; Coulonges, C.; Kuil, L.E.; van Ham, T.J.; Zagury, J.F.; Sieger, D. Gene Expression Profiling Reveals a Conserved Microglia Signature in Larval Zebrafish. Glia 2020, 68, 298–315. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.; Korzh, V.; Strahle, U. Zebrafish Embryos Are Susceptible to the Dopaminergic Neurotoxin MPTP. Eur. J. Neurosci. 2005, 21, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Van Ham, T.J.; Brady, C.A.; Kalicharan, R.D.; Oosterhof, N.; Kuipers, J.; Veenstra-Algra, A.; Sjollema, K.A.; Peterson, R.T.; Kampinga, H.H.; Giepmans, B.N.G. Intravital Correlated Microscopy Reveals Differential Macrophage and Microglial Dynamics during Resolution of Neuroinflammation. Dis. Models Mech. 2014, 7, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Oosterhof, N.; Holtman, I.R.; Kuil, L.E.; van der Linde, H.C.; Boddeke, E.W.G.M.; Eggen, B.J.L.; van Ham, T.J. Identification of a Conserved and Acute Neurodegeneration-Specific Microglial Transcriptome in the Zebrafish. Glia 2017, 65, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.P.; Sheng, D.Z.; Sugimoto, K.; Gonzalez-Rajal, A.; Nakagawa, S.; Hesselson, D.; Kikuchi, K. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev. Cell 2017, 43, 659–672.e5. [Google Scholar] [CrossRef] [PubMed]
- Vijayanathan, Y.; Lim, F.T.; Lim, S.M.; Long, C.M.; Tan, M.P.; Majeed, A.B.A.; Ramasamy, K. 6-OHDA-Lesioned Adult Zebrafish as a Useful Parkinson’s Disease Model for Dopaminergic Neuroregeneration. Neurotox. Res. 2017, 32, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, L.J.; Davies, N.O.; Cavone, L.; Mysiak, K.S.; Semenova, S.A.; Panula, P.; Armstrong, J.D.; Becker, C.G.; Becker, T. Regeneration of Dopaminergic Neurons in Adult Zebrafish Depends on Immune System Activation and Differs for Distinct Populations. J. Neurosci. 2019, 39, 4694–4713. [Google Scholar] [CrossRef] [PubMed]
- Flinn, L.J.; Keatinge, M.; Bretaud, S.; Mortiboys, H.; Matsui, H.; De Felice, E.; Woodroof, H.I.; Brown, L.; McTighe, A.; Soellner, R.; et al. TigarB Causes Mitochondrial Dysfunction and Neuronal Loss in PINK1 Deficiency. Ann. Neurol. 2013, 74, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Edson, A.J.; Hushagen, H.A.; Frøyset, A.K.; Elda, I.; Khan, E.A.; Di Stefano, A.; Fladmark, K.E. Dysregulation in the Brain Protein Profile of Zebrafish Lacking the Parkinson’s Disease-Related Protein DJ-1. Mol. Neurobiol. 2019, 56, 8306–8322. [Google Scholar] [CrossRef] [PubMed]
- Chavali, L.N.M.; Yddal, I.; Bifulco, E.; Mannsåker, S.; Røise, D.; Law, J.O.; Frøyset, A.K.; Grellscheid, S.N.; Fladmark, K.E. Progressive Motor and Non-Motor Symptoms in Park7 Knockout Zebrafish. Int. J. Mol. Sci. 2023, 24, 6456. [Google Scholar] [CrossRef] [PubMed]
- Towns, C.; Richer, M.; Jasaityte, S.; Stafford, E.J.; Joubert, J.; Antar, T.; Martinez-Carrasco, A.; Makarious, M.B.; Casey, B.; Vitale, D.; et al. Defining the Causes of Sporadic Parkinson’s Disease in the Global Parkinson’s Genetics Program (GP2). NPJ Park. Dis. 2023, 9, 131. [Google Scholar] [CrossRef]
- Larbalestier, H.; Keatinge, M.; Watson, L.; White, E.; Gowda, S.; Wei, W.; Koler, K.; Semenova, S.A.; Elkin, A.M.; Rimmer, N.; et al. GCH1 Deficiency Activates Brain Innate Immune Response and Impairs Tyrosine Hydroxylase Homeostasis. J. Neurosci. 2022, 42, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chu, S.; Yang, Y.; Zhang, Z.; Pang, Z.; Chen, N. Neuroinflammatory In Vitro Cell Culture Models and the Potential Applications for Neurological Disorders. Front. Pharmacol. 2021, 12, 671734. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.Y.; Yuan, B.S.; Hou, X.O.; Zhang, X.J.; Pei, C.S.; Ma, Y.T.; Yang, Y.P.; Fan, Y.; Qin, Z.H.; Liu, C.F.; et al. A-synuclein Suppresses Microglial Autophagy and Promotes Neurodegeneration in a Mouse Model of Parkinson’s Disease. Aging Cell 2021, 20, e13522. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Büeler, H. Lack of PINK1 Alters Glia Innate Immune Responses and Enhances Inflammation-Induced, Nitric Oxide-Mediated Neuron Death. Sci. Rep. 2018, 8, 383. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Badanjak, K.; Mulica, P.; Smajic, S.; Delcambre, S.; Tranchevent, L.C.; Diederich, N.; Rauen, T.; Schwamborn, J.C.; Glaab, E.; Cowley, S.A.; et al. IPSC-Derived Microglia as a Model to Study Inflammation in Idiopathic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 740758. [Google Scholar] [CrossRef] [PubMed]
- Rahmoune, H.; Guest, P.C. Chapter 12 Studies of Isolated Peripheral Blood Cells as a Model of Immune Dysfunction. Front. Cell Dev. Biol. 2021, 9, 740758. [Google Scholar] [CrossRef]
- Raulf-Heimsoth, M. T Cell: Primary Culture from Peripheral Blood. In Allergy Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2019; Volume 2020, pp. 17–31. [Google Scholar] [CrossRef] [PubMed]
- Renato Muotri, A.; Consiglio, A.; Simmnacher, K.; Lanfer, J.; Rizo, T.; Kaindl, J.; Winner, B. Modeling Cell-Cell Interactions in Parkinson’s Disease Using Human Stem Cell-Based Models. Front. Cell. Neurosci. 2020, 13, 571. [Google Scholar] [CrossRef]
- Weinert, M.; Selvakumar, T.; Tierney, T.S.; Alavian, K.N. Isolation, Culture and Long-Term Maintenance of Primary Mesencephalic Dopaminergic Neurons From Embryonic Rodent Brains. J. Vis. Exp. 2015, 96, e52475. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, G.; Li, D.; Wang, Y.; Wu, Q.; Shi, J.; Zhang, F. Dual Modulation on Glial Cells by Tetrahydroxystilbene Glucoside Protects against Dopamine Neuronal Loss. J. Neuroinflammation 2018, 15, 161. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Okumura-Torigoe, N.; Miyazaki, I.; Murakami, S.; Kitamura, Y.; Sendo, T. Region-Specific Neuroprotective Features of Astrocytes against Oxidative Stress Induced by 6-Hydroxydopamine. Int. J. Mol. Sci. 2019, 20, 598. [Google Scholar] [CrossRef] [PubMed]
- Goshi, N.; Morgan, R.K.; Lein, P.J.; Seker, E. A Primary Neural Cell Culture Model to Study Neuron, Astrocyte, and Microglia Interactions in Neuroinflammation. J. Neuroinflammation 2020, 17, 155. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Stone, D.K.; Mosley, R.L.; Gendelman, H.E. Nitrated α-Synuclein-Induced Alterations in Microglial Immunity Are Regulated by CD4+ T Cell Subsets. J. Immunol. 2009, 182, 4137–4149. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Scheller, A.; Liu, Y.; Krause, E.; Chang, H.F. Study of Effector CD8+ T Cell Interactions with Cortical Neurons in Response to Inflammation in Mouse Brain Slices and Neuronal Cultures. Int. J. Mol. Sci. 2023, 24, 3166. [Google Scholar] [CrossRef] [PubMed]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-Culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Marxreiter, F.; Krach, F.; Winkler, U. Th17 Lymphocytes Induce Neuronal Cell Death in a Human IPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Samata, B.; Doi, D.; Nishimura, K.; Kikuchi, T.; Watanabe, A.; Sakamoto, Y.; Kakuta, J.; Ono, Y.; Takahashi, J. Purification of Functional Human ES and IPSC-Derived Midbrain Dopaminergic Progenitors Using LRTM1. Nat. Commun. 2016, 7, 13097. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Mao, C.; Fan, L.; Luo, H.; Hu, Z.; Zhang, S.; Yang, Z.; Zheng, H.; Sun, H.; Fan, Y.; et al. Modeling Parkinson’s Disease Using Induced Pluripotent Stem Cells. Curr. Neurol. Neurosci. Rep. 2020, 12, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Moudio, S.; Rodin, F.; Jamal Albargothy, N.; Karlsson, U.; Reyes, J.F.; Hallbeck, M. Exposure of α-Synuclein Aggregates to Organotypic Slice Cultures Recapitulates Key Molecular Features of Parkinson’s Disease. Front. Neurol. 2022, 13, 826102. [Google Scholar] [CrossRef] [PubMed]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Yang, S.M.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell Diversity and Network Dynamics in Photosensitive Human Brain Organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Ormel, P.R.; Vieira de Sá, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia Innately Develop within Cerebral Organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Bardien, S.; Ross, O.A.; Bolognin, S.; Sterneckert, J.; Galet, B.; Cheval, H.; Ravassard, P. Patient-Derived Midbrain Organoids to Explore the Molecular Basis of Parkinson’s Disease. Front. Neurol. 2020, 11, 1005. [Google Scholar] [CrossRef]
- Ao, Z.; Cai, H.; Wu, Z.; Song, S.; Karahan, H.; Kim, B.; Lu, H.-C.; Kim, J.; Mackie, K.; Guo, F. Lab on a Chip Tubular Human Brain Organoids to Model Microglia-Mediated Neuroinflammation. Lab Chip 2021, 21, 2751. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y. Patient-Specific Pluripotent Stem Cell-Based Parkinson’s Disease Models Showing Endogenous Alpha-Synuclein Aggregation. BMB Rep. 2019, 52, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Ponz-Sarvise, M.; Alberto, P.-M.; Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In Vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy Aging and the Blood-Brain Barrier. Nat. Aging 2021, 1, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Elabi, O.; Gaceb, A.; Carlsson, R.; Padel, T.; Soylu-Kucharz, R.; Cortijo, I.; Li, W.; Li, J.Y.; Paul, G. Human α-Synuclein Overexpression in a Mouse Model of Parkinson’s Disease Leads to Vascular Pathology, Blood Brain Barrier Leakage and Pericyte Activation. Sci. Rep. 2021, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Porschen, L.T.; Richter, F.; Gericke, B. Microvascular Blood-Brain Barrier Alterations in Isolated Brain Capillaries of Mice over-Expressing Alpha-Synuclein (Thy1-ASyn Line 61). Neurobiol. Dis. 2023, 187, 106298. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.; Wang, P.; Chan, R.B.; Liu, Z.; Yu, Z.; Liu, X.; Yang, Y.; Zhang, J. Astrocytic VEGFA: An Essential Mediator in Blood-Brain-Barrier Disruption in Parkinson’s Disease. Glia 2022, 70, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zheng, J.; Pettersson, S.; Reynolds, R.; Tan, E.K. The Link between Neuroinflammation and the Neurovascular Unit in Synucleinopathies. Sci. Adv. 2023, 9, eabq1141. [Google Scholar] [CrossRef] [PubMed]
- Özkan, E.; Çetin-Taş, Y.; Şekerdağ, E.; Kızılırmak, A.B.; Taş, A.; Yıldız, E.; Yapıcı-Eser, H.; Karahüseyinoğlu, S.; Zeybel, M.; Gürsoy-Özdemir, Y. Blood-Brain Barrier Leakage and Perivascular Collagen Accumulation Precede Microvessel Rarefaction and Memory Impairment in a Chronic Hypertension Animal Model. Metab. Brain Dis. 2021, 36, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; He, X.Z.; Li, Z.H.; Meng, J.C.; Mao, R.T.; Li, X.; Xue, R.; Gui, Q.; Zhang, G.X.; et al. Interaction Between the Glymphatic System and α-Synuclein in Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; He, R.; Chen, J.; Zhou, X.; Zhou, X.; Liu, Z.; Xu, Q.; Guo, J.F.; Yan, X.X.; Jiang, N.; et al. Neuroimaging Uncovers Distinct Relationships of Glymphatic Dysfunction and Motor Symptoms in Parkinson’s Disease. J. Neurol. 2023, 270, 2649–2658. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Pu, T.; Feng, W.; Lu, M.; Zheng, Y.; Du, R.; Xiao, M.; Hu, G. Blocking Meningeal Lymphatic Drainage Aggravates Parkinson’s Disease-like Pathology in Mice Overexpressing Mutated α-Synuclein. Transl. Neurodegener. 2019, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Shi, L.; Li, Y.; Duan, Q.; Qiu, Y.; Feng, S.; Gao, Y.; Luo, Y.; Ma, G.; Zhang, Y.; et al. The Association of the Glymphatic Function with Parkinson’s Disease Symptoms: Neuroimaging Evidence from Longitudinal and Cross-Sectional Studies. Ann. Neurol. 2023, 94, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Yang, B.; Chen, Y.; Zhao, W.; Li, B.; Jia, W. Enlarged Perivascular Spaces Are Linked to Freezing of Gait in Parkinson’s Disease. Front. Neurol. 2022, 13, 985294. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Berberian, S.A.; Breen, D.P.; Gao, F.; Ozzoude, M.; Adamo, S.; Scott, C.J.M.; Berezuk, C.; Yhap, V.; Mestre, T.A.; et al. Small and Large Magnetic Resonance Imaging-Visible Perivascular Spaces in the Basal Ganglia of Parkinson’s Disease Patients. Mov. Disord. 2022, 37, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, D.d.C.; Lopes Aguiar, C.; Moraes, M.F.D.; Fisone, G. Sleep Disorders in Rodent Models of Parkinson’s Disease. Front. Pharmacol. 2019, 10, 496911. [Google Scholar] [CrossRef] [PubMed]
- Temple, S. Advancing Cell Therapy for Neurodegenerative Diseases. Cell Stem Cell 2023, 30, 512–529. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Culture Models | Advantages | Disadvantages |
|---|---|---|
| Primary microglial cultures | Study of the effect of genetic or toxic alterations associated with PD Controlled conditions, isolated effect | Does not replicate the complexity of the environment or the interaction with other cells of PD |
| IPSCs microglial-derived cells | Less invasive than primary cultures | It is unknown whether it replicates the phenotype of resident microglia |
| Culture of peripheral immune cells | The study of the phenotype of peripheral immune cells Transplantation from PD models to controls or vice versa (in vivo effects) | Require in vitro stimuli to induce cellular activation (e.g., LPS, oxidative stress, etc.) |
| Co-culture models: | They allow for the study of cellular interactions between CNS cells and IS cells under controlled conditions | The structural and environmental complexity is not replicated |
| Microglial + Astrocites + Neurons | ||
| Peripheral IS cells + Brain cells | ||
| IPSCs neurons + IS cells (microglial and macrophages) | ||
| Ex vivo brain slice cultures + immune cells | Better replicate structural complexity and can be co-cultured with immune cells to study their interaction | Does not allow for the study of long-term effects |
| Organoids | Mimic the complexity of brain and cellular interactions Reproduces the genetic background of the animal model or the patient | Does not contain some types of glial or immune cells, needs co-culture with them Requires a long time for proper organoid maturation |
| Animal Model | Technical Approaches | Ref. |
|---|---|---|
| MPTP/mice | Immunofluorescence of GFP+ cells | Depboylu et al., 2012 [148] |
| MPTP/mice | Immunofluorescence | Samantaray et al., 2015 [149] |
| MPTP/mice | Flow cytometry | Yamamoto et al., 2022 [150] |
| LRKK2 transgenic mice | Immunohistochemistry and flow cytometry | Kozina et al., 2018 [151] |
| A53T-α-synuclein injection | Flow cytometry | Karikari et al., 2022 [134] |
| 6-OHDA/mice | TSPO PET scan | Lucot et al., 2022 [136] |
| 6-OHDA/rat | Immunofluorescence | Tentillier et al., 2022 [152] |
| MPTP/monkeys | Immunohistochemistry | Miklossy et al., 2006 [153] |
| Transplantation/monkeys | Immunohistology | Bakay et al., 1998 [119] |
| Zebrafish | Epifluorescence | Zwi et al., 2019 [155] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saponjic, J.; Mejías, R.; Nikolovski, N.; Dragic, M.; Canak, A.; Papoutsopoulou, S.; Gürsoy-Özdemir, Y.; Fladmark, K.E.; Ntavaroukas, P.; Bayar Muluk, N.; et al. Experimental Models to Study Immune Dysfunction in the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 4330. https://doi.org/10.3390/ijms25084330
Saponjic J, Mejías R, Nikolovski N, Dragic M, Canak A, Papoutsopoulou S, Gürsoy-Özdemir Y, Fladmark KE, Ntavaroukas P, Bayar Muluk N, et al. Experimental Models to Study Immune Dysfunction in the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(8):4330. https://doi.org/10.3390/ijms25084330
Chicago/Turabian StyleSaponjic, Jasna, Rebeca Mejías, Neda Nikolovski, Milorad Dragic, Asuman Canak, Stamatia Papoutsopoulou, Yasemin Gürsoy-Özdemir, Kari E. Fladmark, Panagiotis Ntavaroukas, Nuray Bayar Muluk, and et al. 2024. "Experimental Models to Study Immune Dysfunction in the Pathogenesis of Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 8: 4330. https://doi.org/10.3390/ijms25084330
APA StyleSaponjic, J., Mejías, R., Nikolovski, N., Dragic, M., Canak, A., Papoutsopoulou, S., Gürsoy-Özdemir, Y., Fladmark, K. E., Ntavaroukas, P., Bayar Muluk, N., Zeljkovic Jovanovic, M., Fontán-Lozano, Á., Comi, C., & Marino, F. (2024). Experimental Models to Study Immune Dysfunction in the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences, 25(8), 4330. https://doi.org/10.3390/ijms25084330